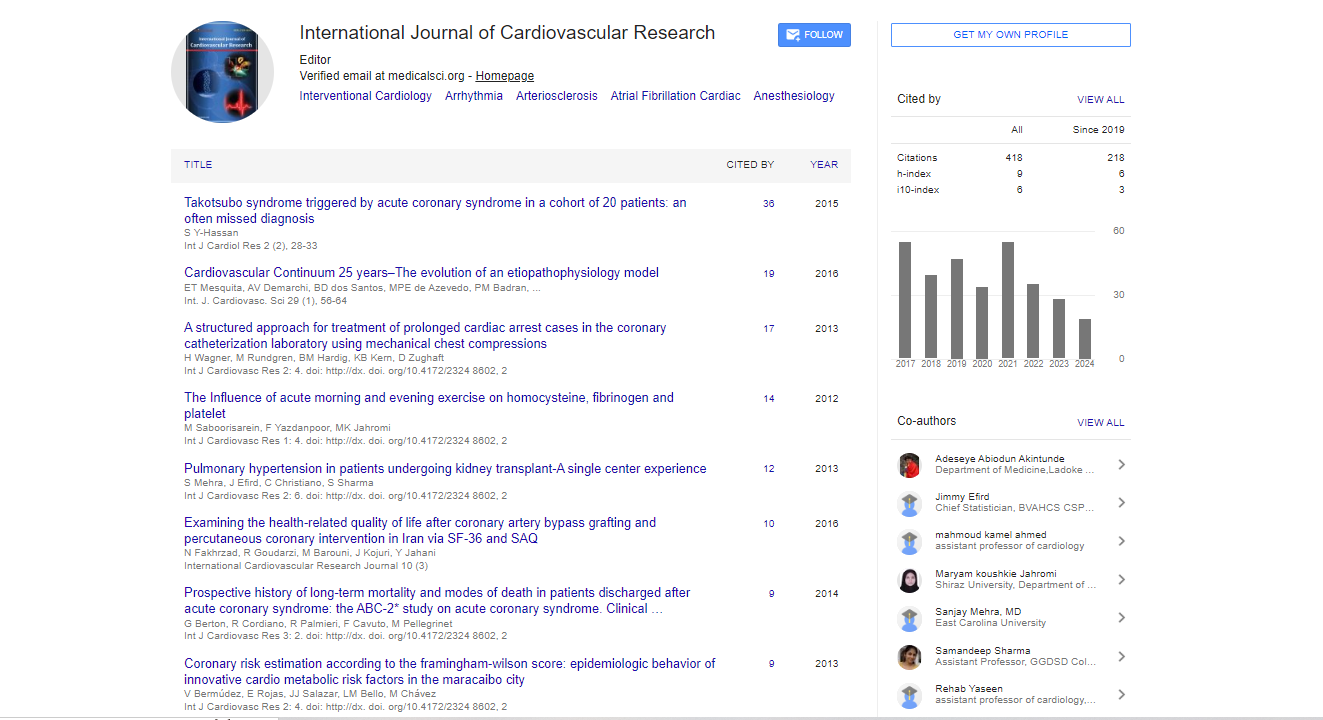
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Editorial, Int J Cardiovasc Res Vol: 1 Issue: 1
Atherosclerosis, Cancer, Wound Healing, and Inflammation - Shared or Parallel Evolution
| Alexandra Lucas* | |
| Division of Cardiovascular Medicine and Rheumatology, University of Florida, USA | |
| Corresponding author : Alexandra Lucas Division of Cardiovascular Medicine and Rheumatology, University of Florida, USA E-mail: alexandra. lucas@medicine.ufl.edu |
|
| Received: May 22, 2012 Accepted: May 25, 2012 Published: May 28, 2012 | |
| Citation: Lucas A (2012) Atherosclerosis, Cancer, Wound Healing, and Inflammation - Shared or Parallel Evolution. Int J Cardiovasc Res 1:1. doi:10.4172/2324-8602.1000e101 |
Abstract
Atherosclerosis, Cancer, Wound Healing, and Inflammation - Shared or Parallel Evolution
Wound healing is a complex process involving inflammatory cell activation and invasion as well as scar tissue deposition (in the form of fibrotic tissue and collagen matrix). With tissue repair there is active cell proliferation designed to heal the damaged tissues. Recent work has suggested that cancer is a form of dysregulated wound healing where inflammatory responses and cellular proliferation goes awry. Additionally other diseases such as the highly prevalent atherosclerotic coronary plaque are hypothesized to be a form of unregulated wound healing.
| Wound healing is a complex process involving inflammatory cell activation and invasion as well as scar tissue deposition (in the form of fibrotic tissue and collagen matrix). With tissue repair there is active cell proliferation designed to heal the damaged tissues. Recent work has suggested that cancer is a form of dysregulated wound healing where inflammatory responses and cellular proliferation goes awry. Additionally other diseases such as the highly prevalent atherosclerotic coronary plaque are hypothesized to be a form of unregulated wound healing. The 'Response to Injury' hypothesis was first presented by Russell Ross as an explanation for atherosclerosis. The basic premise for this theory on the etiology of atherosclerotic arterial disease is that all forms of damage to the arterial wall, whether hypertension, hyperlipidemia, angioplasty injury or transplantation, cause an accelerated injury response with aggressive inflammatory cell responses and cell proliferation, in short an unchecked form of wound healing. Prior to Dr Ross' theory, Earl Benditt postulated that human atheromata were benign intimal tumors. He demonstrated that many atherosclerotic plaques were derived from individual smooth muscle cells using X chromosome inactivation patterns with results that were highly suggestive of a proliferating monoclonal tumour cell. More recent work has now linked excess inflammation with growth and instability of atherosclerotic plaque and with progression of invasive cancers. There are thus now many parallels in developing cancer and atherosclerosis and many close associations between cancer, atheroma and wound healing. These findings raise one basic question – Are carcinogenesis and atherogenesis manifestations of the same initiating disease generating events or simply parallel manifestations of similar pathogenic disease mechanisms, e.g. are these shared or parallel evolutions. |
| The history of our understanding of the pathogenesis of wound healing and inflammation helps in understanding these disorders and the underlying mechanisms of disease. Ilya Ilyich Mechnikov (1845- 1916), a Russian biologist and zoologist who moved to work at the Institute Pasteur in Paris, was the first to discover the existence of innate immunity, also termed inflammation. Although the innate immune system is now known to cure the majority of infections and drives wound healing after injury long before the adaptive immune response becomes active, Mechnikov's initial discovery of this more ancient defense system was viewed with some suspicion and he shared the Nobel Prize for his work in 1908 with Paul Ehrlich. He discovered the inflammatory / innate immune response system by examining wound healing using basic scientific bench work, not translational nor applied research designed to examine a specific disease. In these experiments, he inserted splinters into transparent star fish and noted an immediate rapid massing of inflammatory cells around the splinter. Subsequently similar early cell responses were seen in water fleas infected with microbes. Within minutes he saw a rapid response to these injuries and infections with a swarming of inflammatory mononuclear cells around the offending agents. Another early pioneer was Rudolf Virchow who is attributed with first describing the close association between vascular cell injury, clot formation and inflammation, Virchow's triad of arterial or endothelial injury, stasis of blood flow, and hypercoagulability / thrombosis. Virchow was a remarkable German physician and scientist (1821-1902) who worked as a true Renaissance man, an anthropologist and biologist who improved public health in addition to his work in pathology and science. These two remarkable scientists thus developed the basis for our current understanding of innate immunity and wound healing which are only in recent years becoming recognized as pivotal driving events in progression of wound healing, atheroma, and cancer and no doubt many other disorders. |
| The parallels in these diseases become more evident with each new study into the mechanisms underlying the development of cancer and atherosclerosis. Certainly with atherosclerosis, the original theories suggested either a pure smooth muscle cell proliferative etiology or insudation of lipids, the lipid hypothesis with fat filled foam cells initiating plaque growth. And as mentioned there were early proposals of benign monoclonal tumor cell growth in the intimal layer initiating plaque growth. Current work has however changed these original ideas and the central roles of inflammatory macrophage (foam cells) and T cells in driving cell proliferation, tissue breakdown, and even plaque instability and rupture are now recognized. When plaque is unstable inflammatory macrophage can release metabolizing proteases that disrupt the overlying fibrous cap exposing the inner thrombotic plaque. The inner plaque 'gruel', which is the meaning of the Greek term 'athero', is composed of collagen and fat in addition to invading cells. This gruel is highly thrombotic and exposure to circulating blood leads to sudden thrombotic occlusions, the cause for heart attacks, strokes and peripheral gangrene. These same events also drive progressive dilatation and rupture of the arterial wall, e.g. aneurysm formation which in cases of sudden rupture has very high associated mortality. |
| In atherosclerotic plaque progression and rupture or in aneurysmal dilatation one sees a veritable army of inflammatory macrophage and T cells, adipocytes, smooth muscle cells as well as fibroblasts interacting to either cause arterial damage, plaque rupture or accelerated plaque growth. The damaged endothelium lining the arterial wall and smooth muscle cells also contribute to increased inflammatory responses and atheroma progression. Cell invasion and proliferation is driven by inflammatory cytokines, chemokines and growth factors. The serine proteases in the thrombotic and thrombolytic cascades also interact with inflammatory mediators both causing plaque rupture or hemorrhage or sudden thrombotic arterial occlusions. The coagulation proteases also activate inflammatory cells and mediators in a reciprocal fashion such that inflammation begets clot and clot begets inflammation, much as Virchow predicted. |
| Now current researchers are finding that cancers are also driven by injury and inflammation with similar activations of inflammatory mediators, growth factors and indeed the serine proteases in the coagulation cascades. In fact a tripartite interaction between chronic infections, recurrent inflammation and damage to the colon epithelium is believed to drive cancer progression. Excess inflammation as in inflammatory bowel disease, Crohn's disease and ulcerative colitis, are associated with increased risk of colon cancer. Selected chronic bacterial infections also increase risk of developing tumours in the gastrointestinal tract. Obesity is associated with elevated inflammation and increased risk for early cardiovascular diseases. More recent work has begun to uncover an association between obesity and risk for some cancers, similar to the risk of cardiovascular disease and diabetes in obesity. Cancers throughout the mammalian body are now reported to arise and progress both at sites of injury and scar and in areas with recurrent inflammation and irritation. The same inflammatory responses cells, e.g. Neutrophils, macrophage and T cells are activated and in some cancers appear to drive tumour progression. The close associations of tumor associate macrophage (TAM) and tumor associate neutrophils (TAN) can both initiate cancer cell growth and progression. The recurrent inflammation seen in inflammatory bowel disease is closely associated with increased risk of cancer development and many of the same inflammatory mediators are reported as associated with or driving cancer growth. The cytokine interleukin 6 via STAT signaling, the chemokines that attract cells to sit of injury, caspase associated inflammasome, the thrombotic and thrombolytic serine proteases are present both as markers for tumors as well as potentially driving cancer growth and spread. Many newer therapeutic approaches to cancer have been based upon targeting inflammatory mediator such as chemokies and cytokines and growth factors. The thrombolytic protease, urokinase and tissue type plasminogen activator (tPA and uPA) can activate matrix degrading enzymes (matrix metalloproteinases or MMPs) that in turn can allow cell invasion and increased tumor angiogenesis. The prostaglandins also are active in cancer as well as in atherosclerosis and treatment with aspirin and NSAIDs are associated with altered risk of cancer or plaque rupture and thrombotis. Many of these parallels in cancer and atherosclerosis are also seen in wound healing. These parallels in disease progression in cancer, atherosclerosis, and wound healing were beautifully described in a recent talk by Dr Pual Martin at the Keystone meeting on Carcinogenesis and Inflammation. |
| However, although there are many similar or parallel events driving both diseases, cancers and atheromata, these are not absolute matches for these often similar events. While many of the same pathways and inflammatory responses are seen in atheroma, cancer and wound healing each tumor and each individual has unique modifying events. We have not yet proven whether these are simply similar parallel evolutions of common similar defense responses or whether these diseases represent a shared origin in pathogenic mechanisms or whether these diseases have evolved from a similar set of stimulating events but differing initiators. Thus these observations form a foundation upon which to pursue further studies to examine the origins of these diseases and shared events driven by inflammatory events in wound healing. Further work on these shared events may indeed lead to discovery of newer therapeutic targets shared by many diseases with associated inflammation driven pathogenesis. |