Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Polym Sci Appl Vol: 1 Issue: 1
Improved Catalytic Capabilities of Vanadium Complexes bearing Tridentate Constrained Cyclic β-Enaminoketonato Ligands towards Ethylene (Co)polymerization
| Kaiti Wang1,2, Lei Cui1, Yanguo Li1 and Yuesheng Li3* | |
| 1Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China | |
| 2University of Chinese Academy of Sciences, Changchun Branch, Changchun 130022, China | |
| 3Tianjin Key Lab of Composite & Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300072, China | |
| Corresponding author : Yuesheng Li Tianjin Key Lab of Composite & Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300072, China E-mail: ysli@tju.edu.cn |
|
| Received: February 23, 2017 Accepted: March 13, 2017 Published: March 15, 2017 | |
| Citation: Wang K, Cui L, Li Y, Li Y (2017) Improved Catalytic Capabilities of Vanadium Complexes bearing Tridentate Constrained Cyclic β-Enaminoketonato Ligands towards Ethylene (Co)polymerization. J Polym Sci Appl 1:1. |
Abstract
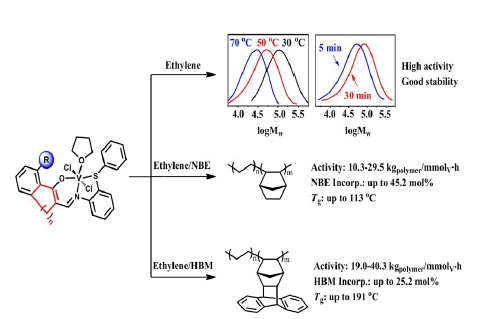
A series of vanadium complexes bearing tridentate constrained cyclic β-enaminoketonato ligands, [RC6H3-(O)C=C(CH2)nCH=NC6H4SPh]VCl2(THF) (2a, n =1, R = H; 2b, n =1, R = C6H5; 2c, n = 2, R = C6H5; 2d, n = 3, R = C6H5), were synthesized and characterized, and the molecular structures of complexes 2b and 2c were further confirmed by X-ray crystal analysis. In the presence of ethyl trichloroacetate as reactivating agent, Et2AlCl activated vanadium complexes produced polyethylenes with unimodal distribution (PDI = 1.4-1.8) in high activities (107-108 gPE/molV•h). Complex 2b bearing phenyl at R position showed higher catalytic activity than complex 2a without bulky R group, and the catalytic properties of these catalysts could be easily tuned by varying the ligand structure and polymerization conditions. These tridentate vanadium complexes showed promising thermal stabilities and did not deactivate in 30 minutes. The copolymerization of ethylene with norbornene (NBE) and exo-1,4,4a,9,9a,10-hexahy-dro-9,10(1′,2′)-benzeno-1,4-methanoanthracene (HBM) were also efficiently catalyzed by these vanadium catalysts, and copolymers possessing high comonomer incorporations (NBE: 45.2 mol%; HBM: 25.2 mol%) and high glass transition temperature (NBE: 113°C; HBM: 191 °C) were obtained.
Keywords: Coordination polymerization; Vanadium catalysts; Polyethylene; Copolymer
Graphical Abstract |
|
 |
|
| Benefitting from the steric effects around the hydroxyl donors, vanadium complexes bearing tridentate constrained cyclic β-enaminoketonato ligands exhibited high catalytic activities and promising stabilities towards ethylene polymerization as well as the copolymerization with cyclic olefin, and produced copolymers possessing high cyclic olefins incorporations and glass transition temperature. | |
Keywords |
|
| Coordination polymerization; Vanadium catalysts; Polyethylene; Copolymer | |
Introduction |
|
| Non-metallocene catalysts for olefin polymerization have experienced celebrated development for decades, and a large number of early and late transition metal catalyst precursors were successfully designed and synthesized to (co)polymerize olefins [1-6]. Among these non-metallocene catalysts, vanadium catalysts exhibit high catalytic activities for producing (co)polymers possessing high molecular weight (MW) and comonomer contents [7-11]. However, the active species may be reduced to low-valent inactive vanadium species and decompose during the polymerizations, which would result in a decline in catalytic activities and the formation of producing polymers with bimodal or multimodal distribution [12-15]. | |
| Introducing tridentate Schiff ligands bearing pendant donors, either the electronic character, steric environment or both of the complexes can be tuned conveniently. This has been proved to be an efficient strategy to improve the stabilities of the vanadium catalysts, as well as other transition metal catalysts. The tridentate Schiff-based titanium catalysts invented by Tang and his colleagues showed outstanding catalytic capabilities, even in copolymerizations of ethylene with polar comonomers [16-19]. Gibson and his coworkers reported that the catalytic activities of chromium catalyst bearing [O, N, N] type tridentate ligands in ethylene polymerization were about one order of magnitude higher than the bidentate analogues [20-22]. Compared with the bidentate salicylaldiminato vanadium complexes, the tridentate complexes reported by our group also showed much improved performances in ethylene polymerization [23,24]. Although the side arm donor showed pronounced stabilization effects on catalytically active species generated from the corresponding catalysts, the catalytic activities of the catalysts decreased because of their influences on the electronic character and steric environment of the active species. | |
| As reported by Morokuma and coworkers, the steric effects around oxygen donor are in favor of lowering the insertion barrier of olefins. Introduction of a bulky group at the ortho position of the oxygen donor, the catalytic activities of the corresponding catalysts would be promoted efficiently [25]. However, in recent years, only a few researches about further improving the steric hindrance around the oxygen donor of tridentate Schiff ligands were reported [26]. The bidentate constrained cyclic β-enaminoketonato ligands developed by us could efficiently enhance the steric hindrance around oxygen donors [27]. Herein, a series of tridentate constrained cyclic β-enaminoketonato ligands and the corresponding vanadium complexes were further synthesized, and these vanadium complexes bearing tridentate constrained cyclic ligands were expected to exhibited both high catalytic activities and improved stabilities. | |
Experimental |
|
| Materials and characterization | |
| All of the air- and/or moisture sensitive compounds were manipulated using standard Schlenk techniques or in an MBraun glovebox under a dry nitrogen atmosphere. All solvents used were purified from an MBraun SPS system. The NMR data of ligands were obtained through a Bruker 400 MHz spectrometer at ambient temperature with CDCl3 or C6D6 as solvent. The NMR data of polymers were obtained through a Bruker 400 MHz spectrometer at 120˚C with o-C6D4Cl2 as solvent. The FT-IR spectra of the complexes were recorded on a Bio-Rad FTS-135 spectrophotometer. Elemental analyses were performed using an Elemental Vario EL spectrometer. Single crystal X-ray analysis were operated with a ω scan mode (185K) on a Bruker Smart APEX diffractometer with CCD detector using Mo Kα radiation (λ=0.71073Å). The molecular weights and the polydispersities of the polyethylene samples were determined at 150°C by a PL-GPC 220 type high-temperature chromatograph equipped with three PL gel 10 μm Mixed-B LS type columns. 1,2,4-Trichlorobenzene (TCB) was employed as the solvent at a flow rate of 1.0 mL/min. The calibration was made by the polystyrene standard Easi Cal PS-1 (PL Ltd.). The melting temperatures and glass transition temperatures were recorded on a Q2000 V24.10 Build 122 DSC differential scanning calorimeter at a rate of 10°C/min. VCl3(THF)3 was purchased from J&K Scientific Ltd., 2-(phenylthio) aniline was charged from Aldrich Chemical and used without any further purification. Ethyl trichloroacetate (ETA) was purchased from Aldrich, dried over calcium hydride at room temperature, and then distilled. Et2AlCl was obtained from Albemarle Corp. Norbornene was purchased from J&K Scientific Ltd., died over Na at 60˚C, and then distilled. Exo-1,4,4a,9,9a,10-hexahy-dro-9,10(1′,2′)- benzeno-1,4-methanoanthracene (HBM) were synthesized according to the literature [28]. Commercial ethylene was directly used for polymerization without further purification. 7-bromoindan-1-one was synthesized according to the literature [29]. The other reagents and solvents were commercially available. | |
| Synthesis of ligands | |
| [C6H4C(OH)=C(CH2)CH=N(C6H4)SC6H5] (1a). 0.7 g (5.0 mmol) of 1-indanone and 0.8 mL (10.0 mmol.) of ethyl formate were added into a slurry of tBuOK (0.9 g, 7.5 mmol) in anhydrous diethyl ether (10.0 mL) at 0˚C, a large amount of white precipitate appeared immediately. The mixture was kept stirring at 0˚C for 30 min, then warmed to room temperature and stirred overnight. The obtained suspension was dealt by Formic acid in ethanol to pH<7. Subsequently, 0.5 mL (5 mmol) of 2-(phenylthio) aniline was added to the solution of β-diketone in ethanol and the condensation reaction was carried out for another 24 h at room temperature. Ligand 1a was obtained as yellow solid after filtration without any further purification (1.52 g, 88.5%). 1H NMR (400 MHz, CDCl3): δ 11.93, 8.04 (d, JHH=11.8, 13.5 Hz, 1H, OH), 7.85, 7.82 (d, JHH=11.8, 13.5 Hz, 1H, Ar-H), 7.61 (dd, JHH=7.6, 1.4 Hz, 1H, Ar-H), 7.56 (dd, JHH=7.7, 1.5 Hz, 1H, Ar- H), 7.53–7.42 (m, 4H, Ar-H), 7.41–7.32 (m, 3H, Ar-H), 7.29–7.19 (m, 3H, Ar-H), 7.19–7.10 (m, 2H, Ar-H), 7.02 (dtd, JHH=18.8, 7.6, 1.1 Hz, 1H, N=CH), 3.65, 3.41 (s, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 193.28 (C-OH), 148.73 (N=CH), 146.95 (N-Ar), 141.24, 140.60 (SAr), 140.20, 137.48, 137.17, 136.38, 135.89, 135.38, 131.61, 130.47, 129.39, 129.24, 128.98, 127.27, 127.06, 126.42, 125.82, 125.68, 123.42, 123.12, 122.75, 121.16, 118.52, 113.58, 112.97, 112.74 (Ar), 109.60 (N=CH-C), 28.42 (CH2); Anal. Calcd. for C22H17ONS: C, 76.94; H, 4.99; N, 4.08; S, 9.34. Found: C, 76.91; H, 4.95; N, 4.03; S, 9.36. | |
| [(C6H5)C6H3C(OH)=C(CH2)CH=N(C6H4)SC6H5] (1b). In a similar way described above, except that 8-phenyl-1-tetralone was added instead of 1-indanone to give 1b as a yellow solid (1.60 g, 76.2%). 1H NMR (400 MHz, CDCl3): δ 11.77, 7.95 (d, JHH=11.6, 13.5 Hz, 1H, OH), 7.63-7.32 (m, 11H, Ar-H), 7.31-7.23 (m, 5H, Ar-H), 7.22-7.10 (m, 3H, Ar-H), 7.00 (dtd, JHH=16.3, 7.5, 1.2 Hz, 1H, N=CH), 3.66, 3.44 (s, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 191.53 (COH), 148.95 (N=CH), 147.29 (N-Ar), 141.36, 141.00 (S-Ar), 140.27, 139.90, 137.52, 137.40, 136.46, 136.14, 135.15, 131.49, 130.69, 129.39, 128.57, 128.37, 127.70, 126.46, 126.40, 126.26, 125.37, 123.66, 121.46, 120.37, 112.42 (Ar), 108.65 (N=CH-C), 29.30 (CH2); Anal. Calcd. for C28H21ONS: C, 80.16; H, 5.05; N, 3.34; S, 7.64. Found: C, 80.13; H, 5.03; N, 3.35; S, 7.66. | |
| [(C6H5)C6H3C(OH)=C(CH2)2CH=N(C6H4)SC6H5] (1c). In a similar way described above, except that 8-phenyl-1-tetralone was added instead of 1-indanone to give 1c as a yellow solid (1.73 g, 79.7%). 1H NMR (400 MHz, CDCl3): δ 12.00, 8.05 (d, JHH=11.8, 13.6 Hz, 1H, OH), 7.60-7.56 (m, 1H, Ar-H), 7.47-7.29 (m, 9H, Ar-H), 7.25- 7.05 (m, 9H, Ar-H), 7.01-6.93 (m, 1H, N=CH), 2.93-2.86 (m, 2H, CH2), 2.66-2.60 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ 186.75 (C-OH), 142.50 (N=CH), 142.47, 142.34 (S-Ar), 141.92 (N-Ar), 137.84, 136.51, 134.46, 132.45, 129.53, 129.50, 129.41, 128.62, 128.30, 127.73, 127.59, 127.11, 126.54, 126.12, 125.28, 125.13, 121.22, 120.43, 112.77 (Ar), 107.09 (N=CH-C), 30.20, 27.34 (CH2); Anal. Calcd. for C29H23ONS: C, 80.34; H, 5.35; N, 3.23; S, 7.40. Found: C, 80.37; H, 5.37; N, 3.21; S, 7.39. | |
| [(C6H5)C6H3C(OH)=C(CH2)3CH=N(C6H4)SC6H5] (1d). In a similar way described above, except that 9-phenyl-1-benzosuberone was added instead of 7-phenyl-1-indanone to give 1d as a yellow solid (1.62 g, 72.3%). 1H NMR (400 MHz, CDCl3): δ 12.07, 8.10 (d, JHH=11.2, 13.8 Hz, 1H, OH), 7.57 (dd, JHH=25.2, 7.6 Hz, 1H, Ar-H), 7.44-7.05 (m, 18H, Ar-H), 6.98 (m, 1H, N=CH), 2.70-2.62 (m, 2H, CH2), 2.50- 2.19 (m, 2H, CH2), 1.87, 1.63 (t, JHH=6.6 Hz, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ 197.44 (C-OH), 142.81 (N=CH), 141.49, 140.88, 139.90, 139.64, 138.31, 137.10, 135.61, 130.41, 129.61, 129.52, 129.12, 128.87, 128.02, 127.28, 126.62, 126.40, 122.77, 121.98, 114.15, 110.29 (Ar), 30.85, 29.86, 27.95 (CH2); Anal. Calcd. for C30H25ONS: C, 80.50; H, 5.63; N, 3.13; S, 7.16. Found: C, 80.47; H, 5.61; N, 3.11; S, 7.13. | |
| Synthesis of complexes | |
| [C6H4C(O)=C(CH2)CH=N(C6H4)SC6H5]VCl2(THF) (2a). A solution of C6H4C(ONa)=C(CH2)CH=N(C6H4)SC6H5 in dried tetrahydrofuran (THF, 15 mL), which was obtained by treating Ligand 1a (0.17 g, 0.5 mmol) with NaH (0.02 g, 0.6 mmol) in THF over 4 hours at room temperature, was added into a solution of VCl3(THF)3 (0.22 g, 0.6 mmol) in tetrahydrofuran (10 mL) drop-wise over 30 min. The reaction mixture was stirred for another 16 h, and filtered to remove NaCl. The resultant black-red solution was concentrated, and the crude product was recrystallized in THF/hexane (v/v=1/3) to give complex 2a as black crystal (0.22 g, 71.0%). FT-IR (KBr pellets): ν/cm-1 3056, 2973, 2874, 1638, 1591, 1561, 1529, 1467, 1446, 1401, 1296, 1271, 1198, 1184, 1166, 1147, 1093, 1024, 1013, 967, 944, 918, 864, 755, 733, 689, 577, 540, 519, 493, 464, 446; Anal. Calcd. for C26H24NO2SVCl2: C, 58.22; H, 4.51; N, 2.61; S, 5.98. Found: C, 58.21; H, 4.51; N, 2.62; S, 5.95. | |
| [(C6H5)C6H3C(O)=C(CH2)CH=N(C6H4)SC6H5]VCl2(THF) (2b) Complex 2b was synthesized according to the same procedure as that of 2a, except that ligand 1b was used in place of 1a (0.22, 68.3%). FT-IR (KBr pellets): ν/cm-1 3078, 3052, 3035, 2970, 2885, 1594, 1561, 1489, 1469, 1442, 1425, 1393, 1297, 1277, 1259, 1197, 1165, 1089, 1076, 1036, 1016, 960, 924, 897, 859, 799, 788, 759, 743, 698, 591, 553, 526, 506, 491, 466, 453, 433; Anal. Calcd. for C32H28NO2SVCl2: C, 62.75; H, 4.61; N, 2.29; S, 5.24. Found: C, 62.77; H, 4.58; N, 2.29; S, 5.28. | |
| [(C6H5)C6H3C(O)=C(CH2)2CH=N(C6H4)SC6H5]VCl2 (THF) (2c). Complex 2c was synthesized according to the same procedure as that of 2a, except that ligand 1c was used in place of 1a (0.24 g, 78.2%). FT-IR (KBr pellets): ν/cm-1 3056, 2946, 2094, 2879, 1644, 1583, 1550, 1481, 1463, 1445, 1416, 1380, 1329, 1281, 1267, 1231, 1212, 1180, 1154, 1092, 1071, 1027, 1016, 998, 951, 871, 856, 831, 802, 783, 758, 747, 732, 718, 696, 685, 649, 587, 554, 514, 467, 449; Anal. Calcd. for C33H30NO2SVCl2: C, 63.26; H, 4.83; N, 2.24; S, 5.12. Found: C, 63.28; H, 4.81; N, 2.22, S, 5.07. | |
| [(C6H5)C6H3C(O)=C(CH2)3CH=N(C6H4)SC6H5]VCl2(THF) (2d). Complex 2d was synthesized according to the same procedure as that of 2a, except that ligand 1d was used in place of 1a (0.24 g, 75.0%). FT-IR (KBr pellets): ν/cm-1 3058, 2932, 2857, 1639, 1579, 1552, 1478, 1463, 1445, 1425, 1369, 1340, 1303, 1270, 1241, 1223, 1209, 1276, 1154, 1092, 1063, 1041, 1023, 1005, 991, 954, 918, 867, 852, 812, 783, 762, 751, 703, 689, 638, 605, 551, 533, 511, 456; Anal. Calcd. for C34H32NO2SVCl2: C, 63.75; H, 5.04; N, 2.19; S, 5.01. Found: C, 63.77; H, 5.01; N, 2.18; S, 5.03. | |
| Ethylene (co) polymerization: A toluene solution of catalysts, comonomers, ethyl trichloroacetate (ETA) and Et2AlCl were prepared in advance, and the polymerizations were carried out in a 150 mL flame-dried Schlenk flask or 200 mL stainless steel reactor with a mechanical stirrer. The reaction flask was thermostatted to the desired temperature and charged with toluene, and then a proper amount of the solution of comonomer, Et2AlCl, ETA and catalyst were injected in sequence into the flask. Subsequently, ethylene was bubbled into the flask at a desired pressure, and the pressure was maintained throughout the reaction. After a desired time, the polymerization was quenched with acidified ethanol, and then poured into 200 mL of acidified ethanol (10% HCl in ethanol). The precipitated polyethylene was collected by filtration, and dried at 60˚C under vacuum till a constant weight. | |
Results and Dicussion |
|
| Synthesis of ligands and complexes | |
| The synthetic routes of ligands and the vanadium complexes are shown in Figure 1 (Scheme 1). 7-phenyl-1-indanone (b), 8-phenyl- 1-tetralone (c) and 9-phenyl-1-benzosuberone (d) were synthesized according to the reported procedures [30,31]. Ligands 1a-1d were then obtained from the previously reported approach by reaction of compounds a-d with 2-(phenylthio)aniline [32]. The tridentate ligands were deprotonated by 1.0 equiv. of NaOH, followed by treating with 1.0 equiv. of VCl3(THF)3 in THF at room temperature. The reaction products were isolated by recrystallization from a mixture of THF and n-hexane (v/v=1/3-1/5) at room temperature. | |
| Figure 1: The synthetic route of the vanadium complexes (Scheme 1). | |
| All of the synthesized ligands were characterized unambiguously by 1H NMR, 13C NMR and elemental analysis. Broad proton signals were observed on the 1H NMR spectra, indicating that these complexes were paramagnetic species. All of the vanadium complexes were airand moisture sensitive and were not suitable for mass spectrometry characterization. Nevertheless, all of these complexes were identified by FT-IR and elemental analysis unambiguously, and the molecular structure of complexes 2b and 2c were further confirmed by single crystal X-ray diffraction analysis. Crystals suitable for crystallographic analysis were obtained from a mixture solution of THF/n-hexane at room temperature. The collection and refinement data are summarized in Table S1, the ORTEP plots are given in Figure 2 and S1, and the selected bond lengths and angles are summarized in Table S2. These complexes are found to adopt a distorted octahedral configuration around the metal center. The tridentate ligands bond with vanadium center via O, N and S donor, and the two chelate rings [V(1)-N(1)-C(8)-C(9)-C(10)-O(1) and V(1)-N(1)-C(1)-C(2)-S(1)] are almost coplanar. The two chloride atoms adopt a trans geometry [Cl(1)-V(1)-Cl(2): 2b, 168.60(3)°; 2c, 169.86(10)°]. The THF molecular occupied at the trans-position of the N donor. The small dihedrals between the planar of bi-chelate rings and the planar of benzocyclanes backbone [torsion angles of O(1)-C(10)-C(11)-C(12): 2b: 1.5°; 2c: 18.6°] indicate that the rotation of the bulky groups ortho to hydroxyl donor are limited, thus the occupations of the groups at R position are constrained close to the O donor and the metal center. Moreover, there were two crystallographically independent molecules in one unit cell of 2c. | |
| Figure 2: Molecular structure of Complex 2b. Ellipsoids are shown with 30% probability. The H atoms are omitted for clarity. | |
| Ethylene Polymerization | |
| In the presence of the co-catalyst Et2AlCl and the reactivating agent ETA, all of these novel complexes exhibited high activities as 107 gPE/molV·h towards ethylene polymerization under atmospheric ethylene pressure, even at elevated temperature. The obtained polyethylenes (PEs) showed unimodal distribution, suggesting the single site behaviors of these catalysts. No signals of branching structures were observed in the 1H NMR spectrum of the PE (Figure S2), indicating that the resultant polymers possessed linear structures [33]. This was further supported by the melting temperatures of 131-135°C, which matched perfectly with that of linear PE. | |
| As reported previously, the steric effects around oxygen donors are beneficial to promoting the catalytic activities of the Schiff-based catalysts [25]. In order to prepare tridentate β-enaminoketonato vanadium catalyst with high activities, a phenyl was introduced into the tridentate ligand at R position and the occupation of the phenyl was constrained by the cyclic skeleton. Compared with complex 2a, complex 2b with a phenyl at R position showed much higher catalytic activity. This result indicated that introducing of bulky groups around the hydroxyl donor of the tridentate β-enaminoketonato ligands were an efficient strategy for improving the catalytic activities of the corresponding vanadium complexes. Moreover, complex 2b also produced PE with higher MW because of its enhanced steric hindrance. The spatial orientation and position of the phenyl were influenced by the skeleton structure. When the members of the cycloalkanes increased, the coordination of ethylene with metal center would be suppressed [27]. Therefore, the catalytic activities of complexes 2b, 2c and 2d decreased in the order of 2b>2c>2d. The chain transfer reaction also will be suppressed by this effect and the molecular weights (MWs) of the obtained PEs followed the opposite trend. | |
| The catalytic properties of these vanadium catalysts were also influenced by the reaction conditions. The complexes showed relative high catalytic activities at elevated temperature, whereas produced PEs possessing low MWs (Entry 1-9, Table 1). Compared with 2a, catalyst 2b showed higher catalytic activities between 30˚C and 70˚C. When the ethylene pressure increased, higher catalytic activity up to 108 gPE/molV·h was achieved and PEs with higher MWs (145-262 kg/ mol) could be obtained (Entry 10-12, Table 1). This result suggested that transfer to the monomer was not the dominant chain transfer pathway in the tridentate vanadium catalytic system. Subsequently, the influence of Et2AlCl feedstock was explored. As shown in Figure 3, the catalytic activity of complex 2c was promoted greatly with the increase of the Et2AlCl feedstock and reached the maximum around a ratio of Al/V=4000, and then decreased slightly as the Et2AlCl feedstock further increased. The MWs of the resultant PEs decreased with the increase of the Et2AlCl feedstock, which proved that transfer to aluminum compounds was the dominant transfer pathway. According to the 1H NMR spectrum of the resultant PE, no signals of terminal vinyl groups were observed (Figure S2), further indicating that the dominant chain transfer pathway was not β-H elimination and transfer to monomers. | |
| Table 1: Ethylene Polymerization catalyzed by the vanadium complexes a | |
| Figure 3: Influences of the Et2AlCl feedstock on ethylene polymerization catalyzed by complex 2c. | |
| Thermal stabilities of the vanadium catalysts | |
| The stabilities against polymerization temperature of these complexes were influenced greatly by the ligand structures. As shown in Figure 4, complexes 2a and 2b showed similar behaviors, the highest activities were achieved at 50˚C, while decreased with further increase of the temperature. However, the catalytic activities of complex 2b were always higher than complex 2a. In comparison with complexes 2a and 2b, complexes 2c and 2d with benzocyclohexane and benzocycloheptane backbone showed improved thermal stabilities. Complex 2b exhibited higher activity than complexes 2c and 2d at the temperature lower than 50˚C, while as the temperature further increased, the activities of complexes 2c and 2d decreased much more slowly than complex 2b. When the polymerization temperature at 70˚C, complex 2c exhibited highest activity among these complexes, and complex 2d showed comparable activity to complex 2b. This phenomenon indicated that the steric hindrance originated from the benzocyclanes backbone was beneficial to improving the stabilities of these complexes. All the complexes bearing bulky R groups showed higher catalytic activities than complex 2a at elevated temperature. Moreover, the PEs obtained at both atmospheric temperature and elevated temperature showed unimodal distribution (Figure S3), further proving that these vanadium catalysts showed promising thermal stabilities. | |
| Figure 4: Thermal stabilities of complexes 2a-2d. | |
| Catalytic performance in prolonged time | |
| The catalytic performance of complexes 2a and 2b in longer period were investigated. As the polymerization time prolonged, the yield of PE increased steadily, and the average activities only decreased slightly. As shown in Figure 5, complex 2a and 2b exhibited an average activity of 4.3 and 7.4 kgPE/mmolV·h, maintaining as about 71.7% and 72.5% of that in the first 5 minutes, respectively. This phenomenon suggested that these vanadium catalysts were still catalytically active toward ethylene polymerization in 30 minutes. Moreover, the obtained PEs exhibited unimodal distribution, and the MWs of the resultant PEs increased with the prolongation of the polymerization time (Figure S4), further proving that the synthesized catalysts did not deactivate in 30 minutes. The lifetimes of complexes 2c and 2d were also evaluated, and similar results were obtained, the typical results were summarized in Table S3. | |
| Figure 5: Lifetime of complex 2a and 2b. The reactions were carried out in 50 mL of toluene at 50°C under atomsphere, and feedstock of catalyst, Et2AlCl and ETA were 0.2 µmol, 0.4 mmol and 0.06 mmol, respectively. | |
| Copolymerization of ethylene with cyclic olefins | |
| These synthesized vanadium complexes also showed high activities toward copolymerizations of ethylene with norbornene (NBE), and produced copolymers with unimodal distribution (Entry 1-6, Table 2). Among these four catalysts, 2b exhibited highest catalytic activity. Although the catalytic activities decreased with the increase of NBE feedstock, it still maintained as 107 kgpolymer/mmolV·h. Furthermore, the complexes with bulky R group also showed higher catalytic activities than complex 2a. When the NBE feedstock was 25 mmol, copolymers with about 28.4-33.1 mol% of NBE incorporations could be obtained by these vanadium catalysts. As the NBE in-feed concentration increased, the NBE incorporations in the resultant copolymers also increased (Entry 2-4, Table 2) Figure 6. According to the 13C NMR spectra of the resultant copolymers, only signals assigned to the isolate NBE units and NBE/E alternating sequences were observed, while the resonances ascribed to repeated NBE insertion were negligible, even in the copolymers possessing high NBE contents [34]. This observation indicated that a type of E/NBE copolymer with approximate alternating structure could be obtained when the NBE incorporation was nearly 50 mol%. Moreover, the glass transition temperature (Tg) of the copolymer with high NBE contents was as high as 113°C. | |
| Table 2: Copolymerization of ethylene with cyclic olefinsa | |
| Figure 6: The 1H NMR spectra of E/NBE copolymers (NBE content: top, 45.2 mol%; bottom, 33.1 mol%). | |
| In order to prepare the E/NBE copolymers with high Tg, the NBE contents in the copolymers have to be promoted to higher than 50 mol%. However, the copolymer with high NBE content showed poor mechanical properties associated with its brittleness. This problem could be well overcome by employing of bulky cyclic olefins [28,35]. These vanadium complexes were investigated as efficient catalysts in the copolymerization of ethylene with HBM, and copolymer with about 25 mol% of HBM incorporation were produced in high catalytic activities at 20 mmol of HBM feedstock (Entry 7-11, Table 2). The copolymers obtained by catalyst 2a possessed comparable HBM incorporations with that produced by 2b, while the MWs were lower (Entry 7 and 8 v.s. Entry 9 and 10, Table 2) Figure 7. Further increase of HBM feedstock was not investigated because of the poor solubility of HBM in toluene. Although the comonomer contents in E/HBM copolymer are lower than that in E/NBE copolymers, the Tg of the resultant copolymers are much higher, which is benefited from the large steric hindrance of HBM. The Tg of the copolymer with about 25 mol% of HBM contents was as high as 190°C. Furthermore, the E/HBM copolymers also possess higher MWs than the E/NBE copolymers. | |
| Figure 7: The 1H NMR spectra of E/HBM copolymers (HBM content: top, 25.2 mol%; bottom, 16.7 mol%). | |
Conclusion |
|
| Several vanadium complexes bearing tridentate constrained cyclic β-enaminoketonato ligands were synthesized and the structures in solid state of complexes 2b and 2c were characterized by single crystal X-ray diffraction. In these complexes, the bulky R groups were constrained by cyclic skeletons close to the hydroxyl donor. Benefiting from the lowering effects of the R groups on the insertion barrier of olefins, these tridentate vanadium complexes showed not only high catalytic activities but also promising thermal stabilities and lifetime toward both the ethylene polymerization and the copolymerization of ethylene with cyclic olefins, and the catalytic properties could be further improved by varying the ligands structures and polymerization conditions. Cyclic olefin copolymers with high comonomer incorporation and high Tg were obtained by these vanadium catalytic systems. In addition, under elevated ethylene pressure, PEs with higher MWs could be obtained and the dominant chain transfer pathway of these vanadium catalytic systems was proved to be transfer to aluminum compounds. | |
Acknowledgement |
|
| The authors are grateful for financial support by China National High-tech R&D Program (No. 2015AA034002). | |
References |
|
|
|