Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Polym Sci Appl Vol: 2 Issue: 1
Poly(Butyl Methacrylate) and Carboxymethyl Dextran Copolymers: Synthesis, Mechanical and Anti-Adhesive Properties
Eléonore Catherine Michel1,2, Pascale Chevallier1, Caroline Loy1, Lucie Lévesque1, Didier Letourneur2 and Diego Mantovani1*
1Laboratory for Biomaterials and Bioengineering, CRC-I, Dept. of Min-Met- Materials Eng. & CHU de Québec Research Center, Laval University, Québec, Québec, G1V 0A6, Canada
2INSERM, U1148, Laboratory for Vascular Translational Science, Institut Galilée, Université Paris 13, Sorbonne Paris Cité, 93430 Villetaneuse, France
*Corresponding Author : Diego Mantovani
Pavillon Adrien-Pouliot, 1065 avenue de la médecine, Québec (Québec) G1V 0A6 Canada
Tel: +1 418 656-2131
E-mail: diego.mantovani@gmn.ulaval.ca
Received: October 10, 2017 Accepted: December 12, 2017 Published: December 15, 2017
Citation: Michel EC, Chevallier P, Loy C, Lévesque L, Letourneur D, et al. (2017) Poly(Butyl Methacrylate) and Carboxymethyl Dextran Copolymers: Synthesis, Mechanical and Anti-Adhesive Properties. J Polym Sci Appl 1:3.
Abstract
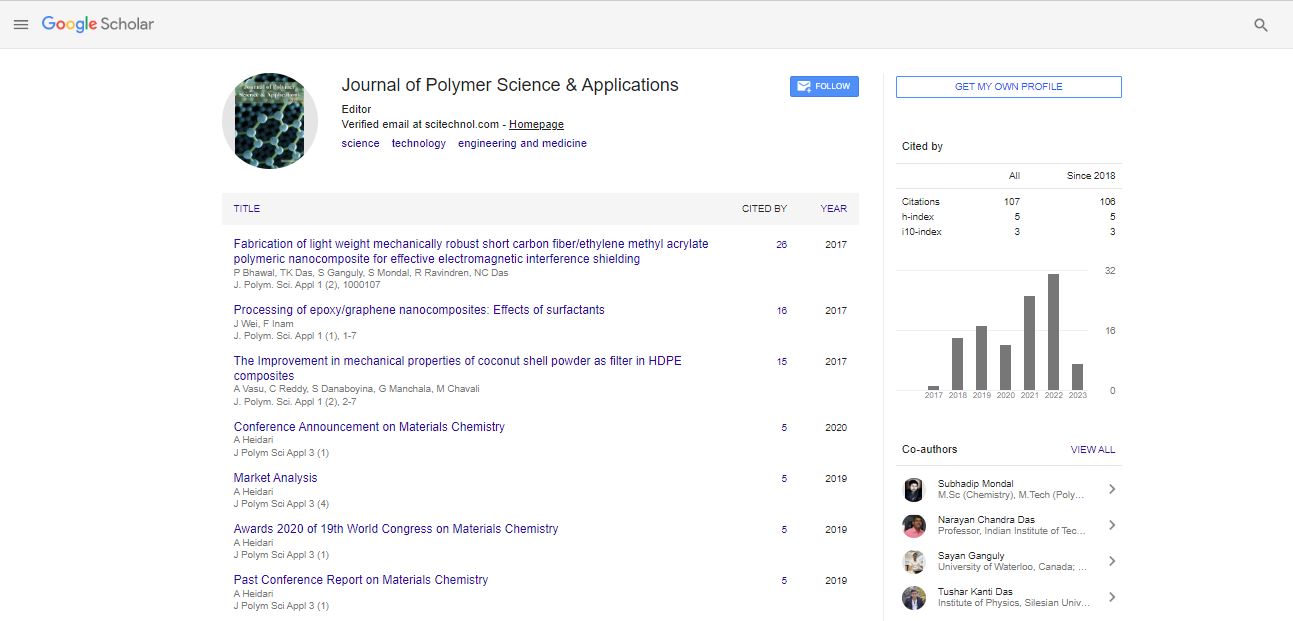
Anti-adhesive and low-fouling properties have a particular interest in some biomedical applications to avoid non-specific cell attachments. In this work, copolymers of carboxymethyl dextran (CMD) and poly(butyl methacrylate) (PBMA) are successfully obtained by free radical copolymerization. The effects of CMD substitution degrees and CMD/BMA ratio on the copolymerization yields are investigated. Copolymers compositions are determined by carbon solid state nuclear magnetic resonance(13C-NMR), Attenuated total reflectance Fourier transform infrared spectra (ATR-FTIR) and by high performance size exclusion chromatography (HPSEC). Results indicate that copolymers are composed of CMD and a majority of PBMA, and PBMA chains are 10-fold longer than CMD ones. Solubility is investigated, and films obtained, in THF/water (90:10), are characterized by X-Ray photoelectron spectroscopy (XPS). Stress relaxation assays evidence flexible and ductile properties. Cell adhesion and proliferation studies with human endothelial cells and 3T3 fibroblasts confirm the absence of toxicity and low adhesion properties. In conclusion, these copolymers show a potential for different applications including coatings for medical devices and biomaterials for which low adhesion properties are required.
Keywords: Carboxymethyl dextran; Poly(butyl methacrylate); Free radical copolymerization; Copolymers; Polymeric film; Mechanical test; Relaxation; Biomaterials
Graphical Abstract
Introduction
The interactions between the surface of the implants and the body are crucial for the clinical success of implants. In some applications, biomaterials and implants are engineered to stimulate a favorable cell or tissue response, in example for vascular prosthesis [1-5]. In other applications, biomaterials and implants are designed to be low-adherent, i.e. to avoid adhesion of proteins and cells. Indeed, non-specific adhesion may lead to issues such as thrombosis, immunological responses or infections [6-9]. To design biomaterials exhibiting low-adhesive properties five major parameters have to be taken into account: surface free energy, electrostatic interactions, steric repulsion, hydration and topography [10-13]. Based on these parameters, a large number of polymers have been investigated so far, mainly for providing cell and protein repellence effect. On the one hand, synthetic polymers, exhibiting smooth surface with low energy such as polyurethanes, poly(dimethyl siloxane), poly(tetrafluoroethylene), and poly(ethylene glycol) for its steric repulsion and hydration parameters, have been reported and tested as bioinert materials for cardiovascular devices, such as endovascular grafts or catheters [8,14]. On the other hand, natural polymers such as polysaccharides might also provide antifouling effects. In this case, these water-soluble polymers could be used as coatings covalently or ionically linked to the substrate, thus providing surfaces with steric repulsion, hydration and electrostatic interaction effects. Carboxymethyldextran (CMD) coatings are particularly interesting because they are known to inhibit non-specific cell and protein adhesion and were found efficient against adhesion of fibroblasts or endothelial cells [15-19]. However, major disadvantages of polysaccharides are their rapid degradation in physiological environment leading to a decrease of their low-adhesive properties and their low mechanical properties. To address these issues, researches were carried out on the effect of mixing synthetic polymers already used in biomedical applications, and polysaccharides to provide materials exhibiting adequate biological and mechanical properties. Thereby, polysaccharides have been bond to oil, resin or plastic polymers such as polyacrylamides polyacrylonitriles, polyacrylates by copolymerization, for diverse applications such as flocculants, bulk coatings or hydrogels [20-28]. In particular, dextran derivatives copolymerized with acrylates have been extensively studied for hydrogel and scaffold fabrication for either drug delivery or tissue engineering applications [3,29-31]. Nevertheless, only a few studies have been conducted on dextran-acrylate based copolymers as possible thin film-coating for biomaterial applications.
Our approach consisted in investigating a novel copolymer based on the reaction of carboxymethyldextran with butyl methacrylate (BMA). Indeed, among synthetic polymers, the poly(butyl methacrylate) (PBMA) is particularly interesting because of its biological and mechanical properties [32-34]. The use of both CMD and PBMA has the potential to lead to a copolymer showing 1) low bio-adhesion and 2) mechanical properties, including elasticity and resistance to deformation. The copolymerization has been thus made using a free radical reaction that easily creates covalent bonds between hydrophilic and hydrophobic polymers [35]. However, such reactions based on production of free radicals are difficult to fine-tune and may lead to many side reactions. To that end, our first challenge was to verify the feasibility of the synthesis and to understand the underlying mechanism of reaction. Then, the syntheses have been characterized by mass balance analyses at each step to study the influence on the copolymerization of the degree of substitution (DS) and CMD/BMA ratio. As a novel amphiphilic material, the copolymer solubility was investigated and no common solvent was found, which challenged liquid state characterization of the copolymers, e.g. proton NMR spectroscopy or size exclusion chromatography. Finally, the copolymers were characterized in solid state by ATR-FTIR and NMR. Films were produced in a mixture of THF/water and tested through tensile relaxation assay and cell adhesion assay.
Experimental
Materials
Dextran (Mw ∼70 kDa) was purchased from TCI Europe (Belgium). Butyl methacrylate (BMA) monomer (99%, d=0.89 g.mL-1, with 10 ppm monomethyl ether hydroquinone as inhibitor) was obtained from Sigma-Aldrich (Steinheim, Germany). Stabilizers were removed by washing monomer (50 mL) successively with an aqueous solution (10 mL) of NaOH (50 g.L-1) and NaCl (200 g.L-1), then three times with deionized water (10 mL). Purified monomers were dried over anhydrous Mg2SO4 and stored under nitrogen at 10°C before use. Ceric(IV) ammonium nitrate (99.5%) was obtained from Acros Organics (Geel, Belgium) and nitric acid (65%) from Chem-Lab (Zedelgem, Belgium). Monochloroacetic acid (Janssen Chimica, Belgium) and others chemicals were analytical reagents grades and used as received.
Graft copolymerization
Before copolymerization, two different carboxymethylated dextrans (CMDs) were synthesized from the native dextran with degrees of substitution (DS) of 0.2 and 0.4, as described in previous work, and referred in this work as CMD 0.2 and CMD 0.4, respectively [36].
The reactions were carried out in a 1 L flask equipped with a mechanical stirrer and a condenser, immersed in a 50 °C thermostatic water bath, under dry nitrogen atmosphere. CMD was dissolved in a 0.2 mol.L-1 HNO3 solution (995 mL) into the flask for 10 min. Then, 0.24 g of Ceric(IV) ammonium nitrate freshly dissolved in 5 mL of 0.2 mol.L-1 HNO3 were added to the medium using a syringe. Exactly 10 min later, 4 mL of BMA were added with a syringe and the mixture was left to react for 2 hours under stirring at 50 °C. After completion of the reaction, the mixture was neutralized by addition of 10 mol.L-1 NaOH and concentrated up to 50 mL with a rotary evaporator at 50 °C. Polymeric macromolecules were recovered by precipitation in icecold ethanol (400 mL). After supernatant elimination, the remaining slurry was transferred into 50 mL centrifuge tubes.
Finally, four types of graft copolymers, CMD-PBMA 1, 2, 3 and 4, were synthesized with two degrees of substitution in CMD and two different weight ratios of CMD/BMA as shown in Table 1.
| Synthesis | HPSECe | |||||
|---|---|---|---|---|---|---|
| CMD | gPBMA | |||||
| Copolymers | DSCMDa | wCMD/wBMA | Yield | hPBMA | Mn | Mn |
| (g/g)b | (%)c | (%)d | (kg.mol-1) | (kg.mol-1) | ||
| CMD-PBMA 1 | 0.2 | 0.07 | 39 ± 9 | 44 ± 9 | 51 | 600– 900 |
| CMD-PBMA 2 | 0.4 | 0.07 | 18 ± 4 | 70 ± 7 | 58 | 600 – 700 |
| CMD-PBMA 3 | 0.2 | 0.3 | 35 ± 3 | 68 ± 4 | 51 | 600 – 900 |
| CMD-PBMA 4 | 0.4 | 0.3 | 30 ± 2 | 60 ± 10 | 58 | 600 – 700 |
a Degree of substitution (DS) of CMD 0.2 and CMD 0.4 previously determined by 1H-NMR.[36]
bInitial weight ratio of CMD (wCMD) and BMA (wBMA) monomers used for the copolymerization.
c Yield of the synthesis defined as the weight of copolymer on the weight of initial CMD and BMA and calculated as follows : WC / (wCMD + wBMA)
dHomopolymer (hPBMA) content formed during the synthesis, determined with the mass loss during Soxhlet extraction, calculated as: (WC+H - WC) / WC+H
e Number average molar mass determined by HPSEC of CMD before copolymerization and ungrafted PBMA (gPBMA) chains from copolymers.
Table 1: Mass balance of the graft copolymerization synthesis in terms of yield and homopolymer content for the 4 initial conditions used.
Extraction of homopolymer and mass balance determination
Ce(IV) ions and unreacted CMD were removed from the previous slurry by alternating vortex mixing and centrifugation washing steps. Briefly, after complete elimination of ethanol, the product was washed twice with 10-2 mol.L-1 EDTA (pH=4.8), to complex and remove ceric ions, and then four times with deionized water. After this first purification step, the products were freeze-dried for 24 h and weighed (noted WC+H).
To remove PBMA homopolymer by-products, the resulting products (2-3 g) were extracted with acetone overnight in a Soxhlet extractor. The pure copolymers were collected in 50 mL centrifuge tubes with 20 mL of acetone. After decantation, the copolymer formed slurry at the bottom of the tube with an acetone supernatant. The copolymer was precipitated by alternating the addition of water droplets with vortex mixing. Remaining acetone was eliminated by washing with deionized water, as previously described, and the resulting product was once more freeze-dried and weighed (noted WC). This procedure results in highly pure copolymers under the form of a fine white powder shape which is useful for further analyses. The yield for each copolymer synthesis was determined as the ratio of the pure graft copolymer weight (WC) to the sum of BMA (WBMA) and CMD (WCMD) weights used for the synthesis. The content of PBMA homopolymer was also calculated as the ratio of the homopolymer weight (WC+H -WC) to the sum of homopolymer and copolymer weights (WC+H).
Side chain separation
Grafted PBMA side chains were removed from the CMD backbone by acid hydrolysis. Briefly, CMD-PBMA (0.5 g) was placed into a flask with 50 mL glacial acetic acid. The flask was fitted with a condenser, placed in an oil bath at 110 °C. The mixture was refluxed for 1 h under stirring to allow the swelling of the grafted side chains. Then, 2 mL of perchloric acid (60%) was added dropwise and the mixture was immediately poured into ice water under vigorous agitation stirring to precipitate the PBMA side chains. The precipitate was recovered by filtration, washed with deionized water and dried at 50 °C under vacuum, weighted and analysed using size exclusion chromatography (cf. Characterization).
Film fabrication
i. Assessment of solubility
Solubility of the copolymers was investigated in various mixtures of water and organic solvents such as: dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), acetone, chloroform and dioxane Table 2. CMD-PBMA (50 mg.mL-1) was dispersed in the solvent in a 15 mL centrifuge tube and placed under magnetic stirring for 1 h, at room temperature. Tubes were then centrifuged to assess the stability. The mixture presenting no phase separation between copolymers and solvent after centrifugation was retained for the film fabrication.
| Solvent/water (%v/v) | 100/0 | 90/10 | 80/20 |
|---|---|---|---|
| DMSO | - | - | - |
| THF | S | D | - |
| Acetone | S | - | - |
| Chloroform | S | - | - |
| Dioxane | S | - | - |
| Water | - | - | - |
Table 2: Behavior of the copolymers in various solvents.
ii. Film molding
In order to perform the mechanical and biological assays, uniform films of copolymers should be obtained. Thus, the four CMD-PBMA (50 mg.mL-1) were dispersed in 4 mL of a mixture of THF and water (90/10 %v/v). The mixtures were then centrifuged at high speed for aggregation removal and the supernatant, a clean and homogeneous dispersion, was kept. This dispersion was then poured in PTFE molds and placed in an atmosphere saturated in THF in presence of CaCl2, overnight. Then, the molds containing the copolymer films were placed under slight vacuum at 40 °C for at least 1 h. Films were gently unmold using tweezers and their thickness were measured with a calliper. Films of PBMA were also made by dissolving ungrafted PBMA chains (50 mg.mL-1) in 4 mL of THF following the previous protocol. Nevertheless, PBMA films were too brittle to be unmolded. For mechanical assay, a dumbbell-shaped mold was used and for biological assay, an evaporating dish.
Characterization
i. Solid state nuclear magnetic resonance (13C-NMR)
Spectra of CMD and CMD-PBMA graft copolymers powders were recorded at 400 MHz with a Bruker Avance spectrometer (Bruker Biospin, Milton, Ontario, Canada) equipped with a magicangle spinning probe head. Spectra were acquired packing samples in 7-mm zirconia tubes and rotated at a frequency of 4 kHz, with a pulse length of 4.5 μs and a recycle delay of 4 s. 4800 scans were acquired for all samples.
ii. Attenuated total reflectance Fourier transform infrared spectra (ATR FTIR)
The spectra of CMD, CMD-PBMA and PBMA homopolymer were recorded by using Attenuated Total Reflectance mode (ATR) on a Cary 660 FTIR (Agilent Technologies, Australia) spectrometer, equipped with a deuterated L-alanine-doped triglycine sulfate (DLa- TGS) detector and a Ge-coated KBr beam splitter. The measurements were recorded in the range 400-4000 cm-1, at a resolution of 4 cm-1, with at least 32 scans from dried powder samples compressed into pellets. Baseline was corrected with multipoint for all spectra and spectra were normalized to the PBMA carbonyl peak at 1724 cm-1 correction, with GRAMS/AI™ Spectroscopy Software (Thermo Scientific™).
iii. Molar mass determination
The molar masses of both CMD and ungrafted side chains of PBMA were determined at room temperature by coupling online a high performance size exclusion chromatograph (HPSEC), a multiangle laser light scattering detector (MiniDAWN TREOS, Wyatt Technology Inc.) and a differential refractive index detector (IOTA 2, Precision Instruments). For CMD analyses, the system was equipped with a Tosoh PWxl guard column and a Tosoh GMPWxl column (1-8000 kDa). Samples (20 mg.mL-1) were eluted in NaNO3-sodium azide (0.15 mol.L-1 and 0.02 %w/v, respectively) filtered solution. For PBMA analyses, the system was equipped with a Tosoh TSKgel Hhr guard column and a Tosoh TSKgel G6000Hhr column (< 4.104 kDa). Samples (20 mg.mL-1) were eluted in filtered acetone. The whole collected data: light scattering and differential refractive index were analysed using the Astra v6.0.6 software package. Molar weights were calculated with dn/dc of 0.142 and 0.124 for CMD and PBMA, respectively, in relation with the mobile phase and its temperature.
iv. Static contact angle (CA)
Measurements were performed on CMD-PBMA films and PBMA coatings (cf. Cell tests – Sample preparations) using a VCA Optima XE system (AST Products Inc.). At least, five drops of deionized water (2 μL) per sample were randomly deposited and pictures were taken after 3 s. The angle value was taken as an average of the number of measures taken for right and left angles.
v. X-Ray photoelectron spectroscopy (XPS)
Surface compositions of CMD-PBMA films and PBMA coatings were assessed by XPS (PHI 5600-ci Spectrometer, Physical Electronics USA). Survey spectra were acquired at a detection angle of 45° using the Kα line of a standard aluminium X-ray source (1486.6 eV) operated at 200 W with charge neutralization, while high resolution spectra were obtained with a standard Mg X-ray source (1253.6 eV) at 150 W without charge neutralization. Detection was performed with a take-off angle of 45° on a 0.5 mm2 area. The spectrometer work function was adjusted to give 285.0 eV for the main C (1s) peak. Curve fittings for high-resolution peaks were determined by means of the least squares minimization procedure employing Gaussian- Lorentzian functions and a Shirley-type background. At least, three measurements per sample were made on three different samples to ascertain the homogeneity and the reproducibility of the surface chemistry.
vi. Mechanical assays
Mechanical tests were carried out on dumbbell-shaped CMDPBMA films with a rectangular section (20 x 9 × 0.09-0.12 mm) with a 100 N load cell mounted on an Instron 5944 single column system section (Instron Corporation, Norwood, MA, USA) at room temperature. The relaxation assay was selected as mechanical test considering its potential to determine the viscoelastic parameters as well as elastic properties of a material, which is not trivial with other assays and which are important parameters for biomaterials under cycling mechanical stress, such as artificial vessel or catheters. Herein, stepwise stress-relaxation tests followed by a tensile test until rupture were performed on each sample to assess its viscoelastic and elongation behaviors’ respectively. The stepwise stress-relaxation test consisted in stretching the sample at a 3% strain (10 mm.min-1) and maintaining the strain constant for 1000 s while the stress decay was monitored. This process was performed repeatedly for each subsequent 3% strain until 27% of total strain. Then, samples were stretched continuously (10 mm.min-1) until fracture. The stress was recorded on Bluehill 3 software (Instron Corporation). The resulting curve express the stress (σ(t)) as a function of time for a constant deformation (ɛ0) (Figure 1). Curve fitting was performed on MatLab 2014 Software (MathWorks) using the Levenberg-Marquardt algorithm to provide the relaxation parameters. The CMD-PBMA copolymers stress response was established to fit a Maxwell–Wiechert model. At least, three samples were tested for each CMD-PBMA copolymer.

Figure 1: A typical stress curve versus time obtained with CMD-PBMA sample.
Cell tests
i. Sample preparations
For cell proliferation assays, CMD-PBMA films were punched in 12 mm-diameter disks that were fixed at the bottom of 24-well plates with Silastic medical adhesive (silicone, type a, Dow Corning Corporation, Auburn, Michigan, USA). Plates were sterilized by immersion in 70% ethanol bath, and washed thereafter with sterile PBS solution.
Cover glass (diam. 12 mm, Fisherbrand) was used as a positive control. PBMA and Silicone were used as comparative controls. PBMA coatings were made from a solution of PBMA (Mw∼337 kg.mol-1, Sigma-Aldrich) in THF (70 mg.mL-1) by depositing 100 μL on a cover glass and placed under same conditioning as CMD-PBMA films. Silicone sheets (87315K72, McMaster-Carr) were punched and fixed as CMD-PBMA samples. All controls were sterilized with ethanol prior biological assay.
ii. Cell culture
Human Umbilical Vein Endothelial Cells (HUVECs) were isolated from an umbilical cord from normal-term pregnancies, kindly provided by the Saint François d’Assise Hospital, with the previous consent of donor mothers. To extract HUVECs, the vein was rinsed with phosphate-buffered saline (PBS, Fair Lawn, New Jersey, USA), filled with trypsin 10x and incubated for 15 minutes at 37 °C. After trypsin removal, the cells were rinsed in PBS, the solution was then recovered and centrifuged at 1000 rpm during 5 min. The supernatant was removed and HUVECs were collected and cultured in a culture flask with M199 culture medium (Gibco by Life Technologies, Grand Island, NY, USA) supplemented with 10% of foetal bovine (Gibco), 1% penicillin/streptomycin (Gibco), Fibroblast Growth Factor-Basic (FGFb, 2ng/mL, Gibco), Epidermal Growth Factor (EGF, 0.5ng/mL, Invitrogen by Life Technologies, Grand Island, NY, USA), L-Ascorbic acid (1μg/mL, Sigma, St Louis, MO), Hydrocortisone (1μg/mL, Sigma), Heparin sodium salt, Grade I-A from porcine (90μg/mL, Sigma), Endothelial Cell Growth Supplement (ECGF, 1μg/mL, Becton Dickinson, Oakville, ON, Canada). This medium will be referred as EC medium thereafter. HUVECs at their third passage were used for the experiments.
NIH-3T3 mouse fibroblast cells line were cultured with DMEM (1x Gibco, 11995-065, Gibco by Life Technologies, Grand Island, NY, USA) culture medium supplemented with 10% of foetal bovine serum, 1% of Penicillin-Streptomycin. This medium will be referred as 3T3 medium thereafter.
HUVECs and 3T3 were collected from the culture flask by the addition of 1X trypsin-EDTA, after the removal of culture medium and rinsing with PBS. After trypsin inactivation with complete medium, the cells were centrifuged for 5 min at 1000 rpm. The resulting pellet was re-suspended in the appropriate volume of medium. For both cytotoxicity and adhesion/proliferation assays, cells were seeded at 40 000 cells/cm2 for HUVECs and 15 000 cells/cm2 for 3T3 fibroblasts.
iii. Cytotoxicity assay
Cytotoxicity test was carried out by indirect contact. Briefly, this test consists to immerge the samples in culture medium and incubate them during 1 and 7 days at 37 °C, and then culture cells with the resulting media. If any toxic elements have been released, the cells will be affected. According to the ISO 10993-5:2009 standard, surfaces were immerged in the desired medium as the ratio between the surface area and the medium volume was 3 cm2/mL. The cytotoxicity assays are performed with the different CMD-PBMA, PBMA, silicone and glass substrates, and with HUVECs (40 000 cells/cm2) as well as 3T3 fibroblasts (15 000 cells/cm2). The HUVEC and 3T3 cells were seeded in 96-well-plates with 200 μL of their respective medium and pre-cultivated for 24 h to obtain optimal layer of cells. The medium was then replaced with extracts (200 μL) after rinsing with PBS. The cells were further incubated for 24 h and their viability was then assessed using resazurin test. Extract medium was replaced by 200 μL of resazurin solution in DMEM (1:10) and let cells metabolize it during 3 h (the oxidation of resazurin led to pink and highly fluorescent resorufin). 150 μL of each resazurin medium were taken and fluorescence was measured at 570 nm using a spectrophotometer ELISA reader (BioRad mod.450, Mississauga, Ontario, Canada). Results are the average of 6 measurements
iv. Cell adhesion and proliferation assay
HUVEC and 3T3 cell adhesion and proliferation were assessed on different CMD-PBMA films as well as on PBMA, silicone and glass samples. Substrates were prepared as described previously. Briefly, 75 μL of each cell suspension were deposited onto the surfaces to be tested, separately. Immediately after, the volume of the deposited cell suspensions was adjusted by addition of culture medium, to completely cover the material surface, then incubated at 37°C for 2 h to allow appropriate cell adhesion. Samples were rinsed with the appropriate culture medium to remove any dead or poorly attached cells and samples were reincubated at 37°C in presence of the appropriate culture medium. After 1, 4 and 7 days, the samples were rinsed with PBS and gently transferred in another 24-well plate to avoid potential bias from possible cells attached to the bottom of the wells. The resazurin assay was conducted as described earlier by adding 750 μL of the 10% resazurin solution in DMEM (1:10) per well and measured for fluorescence at 570nm. The solution was then removed and samples were rinsed 3 times with PBS and were then fixed for 20 min with formaldehyde 3.7% (750 μL). After 3 washing with PBS, cells were permeabilized with saponine (0.1%) and bovine serum albumin (3%) solution in PBS for 10 min. Samples were then immersed in a solution of DAPI (Sigma Aldrich, 32670) (1:3000) and rhodamin-phalloidin TRITC labelled (Sigma) (1:200) in PBS-BSASaponine solution for 1 h. Cells were rinsed three times with Tween 20™ (0.05%) in PBS and mounted with fluoromount Images were recorded using a fluorescence microscope Olympus BX51 (Olympus America Corp., Tokyo, Japan). Results were expressed as the mean of triplicate measurements.
Results and Discussion
Synthesis of CMD-PBMA
Graft copolymers of carboxymethyl dextran with butyl methacrylate (CMD-PBMA) were synthesized by radical polymerization using Ceric(IV) ions as radical initiator. As reported in Table 1, four types of sample were investigated using 2 degrees of substitution of the initial CMD and 2 mass ratio of CMD and BMA monomer. As expected for a radical copolymerization, relatively low yields in CMD-PBMA were obtained (Table 1) for all conditions, mostly because PBMA homopolymers formation competes with CMD-PBMA copolymers. Indeed, after the first step of purification in aqueous medium that eliminates unreacted water-soluble CMD and ceric ions, the resulting product contained the copolymer of CMDPBMA and a large quantity of homopolymer of PBMA (44 to 70 %). The homopolymer was then eliminated by extraction. Note that yields of pure CMD-PBMA made from CMD 0.2 were higher than with CMD 0.4, regardless the ratio CMD/BMA initially used.
The formation of the resulting copolymers was confirmed by ATR-FTIR and solid state 13C-NMR. Figure 2 displays the ATR-FTIR spectra of all 4 CMD-PBMA showing characteristic peaks of PBMA homopolymer, such as stretching vibration of carbonyl (C=O) of the acrylate group at 1724 cm-1, as well as the large band of the hydroxyl (3440 cm-1) vibration from the CMD. The CMD carboxylate C=O vibration were also present on the CMD-PBMA spectra at 1590 cm-1, and their intensity increased with the DS.

Figure 2: FTIR spectra of homopolymer of PBMA (red) and of the 4 copolymers: CMD-PBMA 1 (olive), CMD-PBMA 2 (green), CMD-PBMA 3 (purple) and CMDPBMA 4 (blue). Spectra were normalized to the characteristic band of C=O vibration from acrylate at 1724 cm-1.
Solid state 13C-NMR spectra of all CMD-PBMA were similar as well as spectra of both CMD 0.2 and CMD 0.4. Figure 3 displays a representative 13C-NMR spectrum of each copolymer. PBMA characteristic peaks are clearly evidenced on the CMD-PBMA spectrum with the C=O chemical shift at 178 ppm (d) and the others carbons of PBMA at 10-65 ppm (a-c, e-h). Furthermore, signal of the carbon on the anomeric position in CMD is observed at 98 ppm (1) and the large peak of carbons 2-7 at 73 ppm. Also, on the CMD spectra, carbon of C=O is hardly detected at 178 ppm (8) and C1’at 125 ppm (1’), which is the chemical shift of C1 when the carboxymethyl group is on position 2. Both NMR and FTIR investigations suggest a high content of PBMA in the copolymers CMD-PBMA, due to very low intensity of characteristic peaks of CMD compared to PBMA ones.

Figure 3: Solid state 13C-NMR spectra of CMD and CMD-PBMA copolymer. CMD spectrum is representative of both CMD 0.2 and CMD 0.4. CMD-PBMA spectrum is representative of the four copolymers CMD-PBMA 1, 2, 3 and 4 (Table 1).
To further investigate copolymer compositions, HPSEC analyses were carried out (Table 1). Prior to copolymerization, results obtained with CMD exhibited average molar masses of 50 kg.mol-1 for native dextran, 51 kg.mol-1 for CMD 0.2, and 58 kg.mol-1 for CMD 0.4. Furthermore, the masses obtained were similar to the expected masses calculated from DS obtained in 1H-NMR. After acid hydrolysis, PBMA chains separated from polysaccharides backbone were recovered and also analyzed in HPSEC and 1H-NMR (data not shown). A preliminary study on a commercial PBMA in the same hydrolysis conditions showed that no hydrolysis of the PBMA chains occurred. PBMA side chains of the copolymers were found to be 10- fold longer than CMD ones. No difference was found between the 4 copolymers, and molar masses of ungrafted PBMA were of 600-800 kg.mol-1 in average. The high variation observed in ungrafted PBMA chains lengths may be related to the instability of the radicals involved in the copolymerization reaction and the emulsion process. Ungrafted PBMA chains were also weighted and represent about 80-95% of the weight of the resulting copolymers, for all conditions. This confirms the observation made by FTIR and 13C-NMR analyses, showing that the copolymers were mainly made of PBMA compared to CMD.
Reaction Scheme
The copolymerization reaction scheme is a key parameter regarding the final copolymers compositions. Several articles have been published on the graft copolymerization mechanism of vinyl monomers onto polysaccharides. For all the proposed mechanisms, the principle of radical copolymerization with ceric ions initiator remains the same: the formation of a free radical via a redox reaction in which Ce4+ is reduced to Ce3+ ions and a proton is released (Figure 4a and b). The location of the free radical will determine, inter alia, the position of grafted PBMA chains on the CMD backbone. Literature reports that the free radicals are mainly formed on the polysaccharide chain. To that end, three possible sites are considered: the hemiacetal of the reducing end-point, glycols, and hydroxyls (Figure 4b) [22,37-42]. The end-point hemiacetal is in constant equilibrium with the aldehyde form, giving to the terminal unit high reducing properties. Therefore, the terminal aldehyde is much more reactive than glycols and hydroxyls to oxidants, and thus favored for free radical formation [43]. To investigate if the terminal aldehyde (HCO) is the only site for radical formation, then it has been reduced into alcohol (OH) on CMD chains and copolymerizations were carried out under the same conditions as CMD-PBMA 3 and 4 (higher CMD/BMA ratio). Even with the aldehyde blocked, the copolymerization occurred and the resulting copolymers presented similar compositions as CMD-PBMA 3 and 4 (data not shown). Hence, the reducing end-point (HCO) is not the only active site for radical formation and when blocked, radical formation occurred on other sites. Besides, glycols and OH are much more numerous than the HCO, which is only one per CMD chain, so stands a number-reactivity competition. Hydroxyls in position 2 are known to be more reactive than the other hydroxyls on the dextran unit, and the low yields observed previously for CMD-PBMA 2 and 4 made from CMD 0.4 compared to CMD 0.2, might be explained by the lower availability of this position due to carboxymethylation and suggest that OH and HCO sites compete for radical formation [44].

Figure 4: Suggested reaction scheme for CMD-PBMA copolymerization. Reaction between CMD chains and ceric ions (a) creates radicals on CMD chains in 3 different sites (hydroxyl, glycol, hemiacetal) and free radicals in solution (b). Radicals react with BMA monomers to produce macroradicals of growing homopolymer chains (c) and their recombination end forming majorly copolymer CMD-PBMA and homopolymer of PBMA (d).
After determining the possible location of the radical on the CMD chains, it is of interest to understand how PBMA chains were polymerized and grafted to CMD (Figure 4c). The most common mechanism described in the literature suggests that the free radical formed on the polysaccharide (Polysa.) chain reacts with the vinyl function of the monomer (M) and create the chain reaction for propagation, then termination occurs by recombination, as seen below [24,27,39,41,45]:
Initiation:
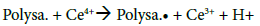
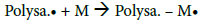
Propagation:
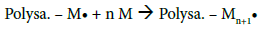
Termination:
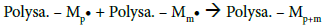
This mechanism suggests high frequency of grafting and short PBMA chains. However, average calculations from ungrafted PBMA molar masses and copolymers weights suggest a low frequency of PBMA grafting on CMD chains which would be 1 or 2 PBMA chains per one CMD chain. Besides, HPSEC analyses (data not shown) demonstrated that PBMA homopolymer chains and ungrafted PBMA side chains exhibit similar average molar masses. These observations are in good agreement with the mechanism proposed by Gaylord et al., considering that graft copolymerization occurs mainly by the termination of growing homopolymer on the polysaccharide backbone [37,38,46-48]. Ceric ions may create free radicals present in the solution that would initiate the homopolymerization [49]. The resulting mechanism would be thus as follows:
Initiation:
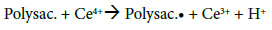
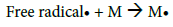
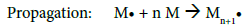
Termination:
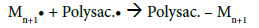
Both mechanisms may occur (Figure 4). However, CMD-PBMA copolymers are suggested to be mainly the result of the growing PBMA chains termination on the hemiacetal or hydroxyl (position 2) of the CMD chain, resulting in a block copolymer structure rather than a comb structure.
Solubility and Films of CMD-PBMA
CMD-PBMA copolymers solubility was investigated in order to produce polymeric films. As seen previously, CMD-PBMA is mainly made of PBMA chains. Therefore, known solvents of PBMA were firstly tested and then other available organic solvents were also assessed. Due to the hydrophilic CMD chains in the copolymers structure, mixture of organic solvents with water in different ratio was also tested (Table 2). No solvents were found to dissolve both CMD and PBMA chains. CMD-PBMA 1, 2, 3 and 4 were found to behave similarly in the tested solvents and mixtures. However, a swelling of chains was observed in THF, acetone, chloroform and dioxane but in mixtures of these solvents with water, the copolymers remained precipitated with the exception of THF. Indeed, when 10% of water was added to THF, copolymers also swelled. The resulting slurry of copolymers in solvents and mixtures of solvents all appeared as colloidal dispersion. Nevertheless, after centrifugation, only THF/water 90:10 presented no phase separation between the slurry of copolymer and the solvent. CMD-PBMA formed a colloidal dispersion only in that mixture.
CMD-PBMA films were made and their chemical compositions determined using XPS analysis (Table 3). PBMA alone was also analyzed for comparison. Our results show no significant differences between each copolymers and chemical compositions were similar to PBMA with a carbon-oxygen ratio around 4. Indeed, considering the low quantity of CMD in the copolymer and the similar composition of CMD and PBMA in terms of chemical bonds and elements, no differences were thus detected both in survey composition as well as in high resolution spectra of C1s and O1s.
| Film | %C | %O | C/O | CA (°) |
|---|---|---|---|---|
| CMD-PBMA | [285.0 eVa] | [532.0 eVc] | ||
| (286.5 eVb) | (533.4 eVb) | |||
| {288.8 eVc} | ||||
| 1 | 80.4 ± 0.4 | 19.6 ± 0.4 | 4.1 ± 0.1 | 84 ± 3 |
| [59.5 ± 1.6] | [9.7 ± 0.9] | |||
| (13.1 ± 1.7) | (10.0 ± 0.6) | |||
| {7.7 ± 0.8} | ||||
| 2 | 79.4 ± 1.4 | 20.6 ± 1.4 | 3.9 ± 0.3 | 80 ± 1 |
| [57.2 ± 2.9] | [9.9 ± 1.6] | |||
| (13.9 ± 1.7) | (10.7 ± 1.0) | |||
| {8.3 ± 0.3} | ||||
| 3 | 79.9 ± 2.4 | 20.1 ± 2.4 | 4.0 ± 0.6 | 82 ± 3 |
| [61.2 ± 5.0] | [10.2 ± 1.7] | |||
| (11.4 ± 1.6) | (9.9 ± 1.4) | |||
| {7.2 ± 1.6} | ||||
| 4 | 77.8 ± 2.1 | 22.2 ± 2.1 | 3.5 ± 0.4 | 82 ± 2 |
| [58.8 ± 3.6] | [12.0 ± 2.9] | |||
| (11.4 ± 1.7) | (10.0 ± 1.1) | |||
| {7.5 ± 0.4} | ||||
| PBMA | 79.7 ± 0.5 | 20.3 ± 0.5 | 3.9 ± 0.1 | 88 ± 2 |
| [58.4 ± 1.9] | [9.3 ± 0.7] | |||
| (12.7 ± 1.5) | (11.0 ± 0.4) | |||
| {8.6 ± 0.4} |
Table 3: XPS characterizations and contact angles (CA) of CMD-PBMA films and PBMA coating.
Nevertheless, the addition of CMD to PBMA in the copolymer decreased the hydrophobicity of the surfaces, exhibiting contacts angle from 88 to 80-84°. This suggests that the CMD chains were exposed at the surface even if no differences were observed in XPS analysis.
Mechanical Properties
Mechanical properties of the CMD-PBMA copolymers were assessed with stepwise stress-relaxation and tensile tests. For small deformations, the copolymer stress relaxation observed could be correlated to a linear viscoelastic behavior due to the ability of polymer chains to reorganize [50-51]. By using the current model described in experimental part, at infinite time only the spring contributes to the stress response meaning only the elastic part of the copolymer, and the elastic moduli (E0,i) was determined for each relaxation step (i). Therefore, for each copolymer, the stress at infinite time (σ0,i = E0,iɛ0,i) of each successive relaxation assay (i) at constant deformation (ɛ0,i), was reported on a stress-strain curve (σ0,i(ɛ0,i)) in function of each ɛ0,i and a linear regression has been processed. The resulting slope corresponds to the linear modulus (EL) of the copolymer (Table 4). CMD-PBMA copolymers were elastic with a linear moduli of 1.5-2 MPa. Furthermore, copolymers are stretchy materials showing deformations up to 60-100% of their initial length before rupture. CMD-PBMA 1 showed a higher elongation to rupture (er = 1.1) compared to the others (er = 0.6). Strain to rupture measurements were performed only on clean-cut fractures and not on eventual film defects, e.g. bulk micro-cuts caused by fabrication. However, elongation responses strongly depend on the film fabrication and test conditions, which may explain the high deformation of CMD-PBMA 1. It should also be underlined that all copolymers films recovered their initial dimensions 12 hours after testing.
| CMD-PBMA | EL (MPa) | er |
|---|---|---|
| 1 | 2.06 ± 0.60 | 1.15 ± 0.13 |
| 2 | 1.51 ± 0.03 | 0.62 ± 0.06 |
| 3 | 2.26 ± 0.11 | 0.58 ± 0.05 |
| 4 | 1.59 ± 0.34 | 0.61 ± 0.04 |
| PBMA | Too brittle to be removed from the mold | |
Table 4: Linear modulus (EL) and elongation to rupture (er) of CMD-PBMA films determined from stress-relaxation assays.
Results evidenced an improvement of mechanical behavior, in terms of elasticity and toughness properties for the copolymers compared to PBMA. Indeed, PBMA films made from ungrafted PBMA chains and processed in the same conditions appeared to be too brittle to be removed from their mold, therefore no tensile test could be performed. In the copolymers, the elasticity is highly improved compared to PBMA. For comparison, a PBMA film with a thickness of hundreds of nanometers presented a Young modulus of 1 270 MPa measured by nanoindentation [52]. Furthermore, the mechanical behavior of the copolymers could be compared to silicone rubber, a widely used material in biomedical applications, which can present a Young modulus of 1-5 MPa and elongation between 100- 800% [53,54].
Cell Adhesion
The biological properties of the copolymer films were assessed through human fibroblast and endothelial cell cultures. As seen previously, all copolymers exhibited similar chemical composition after XPS analyses and similar contact angles. Therefore, only CMDPBMA 3 and 4 were selected for biological testing as they were synthesized with the higher ratio of CMD/BMA and the two different DS of 0.2 and 0.4, CMD-PBMA 3 and 4, respectively. Hence, the influence of the CMD content and the DS in the copolymer have been investigated towards cell adhesion and compared with PBMA. Silicone was taken as control.
Prior adhesion, cytotoxicity of sample extracts was investigated on HUVECs and 3T3 fibroblasts. As shown in Figure 5, copolymers samples as well as PBMA and silicone controls presented cell viability higher than 80% for both HUVEC and 3T3 cells and for all extraction times (1 and 7 days). Results are expressed as a percentage of viability of cells cultivated with the medium which was in contact with glass coverslip, known as a non-cytotoxic control, during 1 and 7 days. According to ISO 10933-5, a reduction of cell viability by less than 20% is considered a non-cytotoxic effect. Hence, no cytotoxicity has been presented by the copolymers samples neither by the PBMA and silicon controls.

Figure 5: Cytotoxicity assay by resazurin absorbance after 1 and 7 days of extraction medium. Cell viability after 24 h of culture expressed as a percentage of the viability of cells in the control: cells cultivated with extracted medium from glass cover-slip immersion. The dashed line shows 80% cell viability (viability > 80%: no cytotoxicity).
Adhesion and proliferation of HUVECs and 3T3 fibroblasts were then investigated on CMD-PBMA 3 and CMD-PBMA 4 as well as on PBMA and silicone, as polymeric controls, and on glass, as a positive control. Cell adhesion (Figure 6) on all samples was low (2%) as compared to glass for both HUVEC and 3T3 cells, and at all times (1, 4 and 7 days). This means that none or very few cells were found on copolymers as well as on PBMA and silicone. Low HUVEC adhesion was observed after 1 day of contact between cells and surfaces for all samples (Figure 6.A.). HUVECs poorly attached and did not proliferate. No significant differences were observed between copolymers and PBMA and silicone. Fluorescence microscopy micrographs have been taken (Figure 7). HUVECs were mostly found in islets. An example of islets is showed on Figure 7. Cells were not homogeneously spread and nuclei were damaged, unable to further proliferate (no cells observed after 4 and 7 days Figure 6 A.), whereas on glass cells were homogeneously spread after 1 day and proliferation has been observed up to 4 and 7 days. CMD-PBMA 3, CMD-PBMA 4, PBMA and silicone only presented remains of dead cells on the surface after 4 and 7 days.

Figure 6: HUVEC (A) and 3T3 fibroblasts (B) presence after 1, 4 and 7 days of culture (D1, D4 and D7) on polymeric surfaces. Cell adhesion expressed as a percentage of cells versus the control on glass.

Figure 7: Fluorescent micrographs of HUVEC (top panel) and 3T3 cells (bottom panel) after 4 days of culture. From left: Representative micrographs of Glass, both PBMA and Silicone (Sil.) and CMD-PBMA 3 and 4 (CMD-PBMA). Right panel: HUVEC islets at day 1 and fibroblast clusters on polymeric surfaces. Nuclei are stained with DAPI (blue) and cell cytoskeleton (actin filaments) with rhodamine-phalloïdine (red). Magnification x 20.
As for 3T3 fibroblasts, no cell adhesion on copolymers has been observed after 1 day (Figure 6.B.). Fluorescence images exhibited some round-shaped cells attached after 1 day for all samples (data not shown). Furthermore, after 4 and 7 days, it can be observed that fibroblasts formed clusters of different sizes on copolymers (Figure 7). It is suggested that few cells attached after 1 day but expressed a very low metabolic activity related to a surviving state, which explains why no adhesion was observed in the resazurine assay. Then, up to 4 and 7 days, fibroblasts unable to adhere to the copolymers surfaces, gathered in clusters to survive. Clusters presented a filaments network in which cells may be able to survive for a while before dying. This explains the viability and the high variations observed after 4 and 7 days for CMD-PBMA 3 and 4. For the silicon and PBMA controls, no cells or not well-spread and isolated ones was observed up to 4 and 7 days, whereas on glass control cells growth occurred until forming a layer after 7 days.
Conclusions
Copolymers of CMD-PBMA obtained from CMD with a DS of 0.2 and 0.4 and with two different ratios of CMD/BMA, 0.07 and 0.3 (w/w), were successfully synthesized. The increase of the degree of substitution from 0.2 to 0.4 seemed to hindrance the copolymerization reaction and higher CMD/BMA ratio increase the yield of the synthesis, which is related to reaction kinetics. CMDPBMA copolymers characterized by ATR-FTIR, 13C-NMR and XPS exhibited similar compositions for all conditions with 80-95 % of PBMA in weight. Furthermore, side grafted PBMA chains were found to be 10-fold longer than CMD, as evidenced by HPSEC, and are grafted to CMD backbone with a low frequency, thus leading to a block copolymer structure. The copolymerization mechanism could therefore imply the termination of growing PBMA homopolymer chains on CMD backbone. Despite the small content of CMD in the copolymer films, the resulted contact angles decreased and the mechanical properties were significantly improved, if compared to PBMA homopolymer. Indeed, CMD-PBMA films appeared elastic and highly stretchable, and able to deform up to 100% of deformation. CMD-PBMA films presented non-cytotoxic and low cell adhesive properties, since fibroblasts or endothelial cells did not adhered. The degree of substitution in CMD had no influence on the biological behavior of the copolymers. Finally, the mechanical and low adhesive properties make the CMD-PBMA copolymers as interesting candidates for medical devices. For example, catheters require elastic, resistant materials able to exhibit anti-adhesive properties towards biological components to avoid encrustation, thrombosis or infection, which are the major factors related to the failure of catheters use.
Acknowledgements
This work was supported by Inserm-FRSQ, FP7 European program PRESTIGE, the Natural Science and Engineering Research Council of Canada, the Quebec Ministry for International Relations, the Centre Franco-Québecois de Coopération Universitaire, Centre d’Études et de Recherche sur les Matériaux Avancés (CERMA-ULaval), the Centre Franco-Québecois de Coopération Universitaire, and the Research Centre of CHU de Québec. The authors would like to express their gratitude to Pierre Saboural, Angélika Kroshko, Bernard Drouin, Pierre Audet and René C.-Gaudreault for their expertise and technical support, as well to all donors of human umbilical veins.
References
- Thalla PK, Fadlallah H, Liberelle B, Lequoy P, De Crescenzo G, et al (2014) Chondroitin Sulfate Coatings Display Low Platelet but High Endothelial Cell Adhesive Properties Favorable for Vascular Implants. Biomacromolecules 15: 2512-2520.
- Hersel U, Dahmen C, Kessler H (2003) RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 24: 4385-4415.
- Möller S, Weisser J, Bischoff S, Schnabelrauch M (2007) Dextran and hyaluronan methacrylate based hydrogels as matrices for soft tissue reconstruction. Biomol Eng 24: 496-504.
- Bacakova L, Filova E, Parizek M, Ruml T, Svorcik V (2011) Modulation of cell adhesion, proliferation and differentiation on materials designed for body implants. Biotechnol Adv 29: 739-767.
- Thierry B, Winnik FM, Merhi Y, Silver J, Tabrizian M (2003) Bioactive Coatings of Endovascular Stents Based on Polyelectrolyte Multilayers. Biomacromolecules 4: 1564-1571.
- Govindarajan T, Shandas R (2014) A Survey of Surface Modification Techniques for Next-Generation Shape Memory Polymer Stent Devices. Polymers (Basel) 6: 2309-2331.
- Lowe HC, Oesterle SN, Khachigian LM (2002) Coronary in-stent restenosis: Current status and future strategies. J Am Coll Cardiol 39: 183-193.
- Bachleda P, Utikal P, Kalinova L, Kocher M, Cerna M, et al (2010) Infectious complications of arteriovenous eptfe grafts for hemodialysis. Biomed Pap 154: 13-19.
- Ronco C (2006) Is the peritoneal catheter a barrier for a wider utilization of PD? Let’s go ahead with research and development! Int J Artif Organs 29: 1.
- Chen S, Li L, Zhao C, Zheng J (2010) Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer (Guildf) 51: 5283-5293.
- Katsikogianni M, Missirlis YF (2004) Concise review of mechanisms of bacterial adhesion to biomaterials and of techniques used in estimating bacteria-material interactions. Eur Cell Mater 8: 37-57.
- Murphy-Ullrich JE (2001) The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest 107: 785-790.
- Vladkova TG (2010) Surface Engineered Polymeric Biomaterials with Improved Biocontact Properties. Int J Polym Sci 2010: 1-22.
- Curtis J, Klykken P (2008) A Comparative Assessment of Three Common Catheter Materials. Dow Corning 1-8.
- McLean KM, Johnson G, Chatelier RC, Beumer GJ, Steele JG, et al (2000) Method of immobilization of carboxymethyl-dextran affects resistance to tissue and cell colonization. Colloids Surfaces B Biointerfaces 18: 221-234.
- Massia SP, Stark J, Letbetter DS (2000) Surface-immobilized dextran limits cell adhesion and spreading. Biomaterials 21: 2253-2261.
- McArthur SL, McLean KM, Kingshott P, St John H a. W, Chatelier RC, et al (2000) Effect of polysaccharide structure on protein adsorption. Colloids Surfaces B Biointerfaces 17: 37-48.
- Monchaux E, Vermette P (2008) Cell adhesion resistance mechanisms using arrays of dextran-derivative layers. J Biomed Mater Res Part A 85A: 1052-1063.
- Hadjizadeh A (2010) Acetaldehyde plasma polymer-coated PET fibers for endothelial cell patterning: Chemical, topographical, and biological analysis. J Biomed Mater Res B Appl Biomater 94: 11-21.
- Sand A, Yadav M, Behari K (2010) Preparation and characterization of modified sodium carboxymethyl cellulose via free radical graft copolymerization of vinyl sulfonic acid in aqueous media. Carbohydr Polym 81: 97-103.
- Biswal DR, Singh RP (2004) Characterisation of carboxymethyl cellulose and polyacrylamide graft copolymer. Carbohydr Polym 57: 379-387.
- Wang L-Q, Tu K, Li Y, Zhang J, Jiang L, et al (2002) Synthesis and characterization of temperature responsive graft copolymers of dextran with poly(N-isopropylacrylamide). React Funct Polym 53: 19-27.
- Zohuriaan-Mehr M (2005) Modified CMC: part1-optimized synthesis of carboxymethyl cellulose-g-polyacrylonitrile. Iran Polym J 14: 131-138.
- Pourjavadi A, Mahdavinia GR, Zohuriaan-Mehr MJ, Omidian H (2003) Modified chitosan. I. Optimized cerium ammonium nitrate-induced synthesis of chitosan-graft-polyacrylonitrile. J Appl Polym Sci 88: 2048-2054.
- Elizalde-Peña EA, Flores-Ramirez N, Luna-Barcenas G, Vásquez-García SR, Arámbula-Villa G, et al (2007) Synthesis and characterization of chitosan-g-glycidyl methacrylate with methyl methacrylate. Eur Polym J 43: 3963-3969.
- Worzakowska M, Grochowicz M (2015) Effect of some parameters on the synthesis and the physico-chemical properties of new amphiphilic starch-g-copolymers. Carbohydr Polym 130: 344-352.
- Rahman L, Silong S, Zin WM, Rahman MZA, Ahmad M, et al (2000) Graft copolymerization of methyl acrylate onto sago starch using ceric ammonium nitrate as an initiator. J Appl Polym Sci 76: 516-523.
- Kadokawa J, Saitou S, Shoda S (2005) Preparation of alginate-polymethacrylate hybrid material by radical polymerization of cationic methacrylate monomer in the presence of sodium alginate. Carbohydr Polym 60: 253-258.
- Lévesque SG, Lim RM, Shoichet MS (2005) Macroporous interconnected dextran scaffolds of controlled porosity for tissue-engineering applications. Biomaterials 26: 7436-7446.
- Derkaoui SM, Avramoglou T, Barbaud C, Letourneur D (2008) Synthesis and Characterization of a New Polysaccharide- graft -polymethacrylate Copolymer for Three-Dimensional Hybrid Hydrogels. Biomacromolecules 9: 3033-3038.
- Kim S-H, Chu C-C (2000) Synthesis and characterization of dextran-methacrylate hydrogels and structural study by SEM. J Biomed Mater Res 49: 517-527.
- Chan C-K, Chu I-M (2001) Phase behavior and molecular chain environment of organic-inorganic hybrid materials based on poly(n-butyl methacrylate-co-(3-(methacryloxypropyl)) trimethoxysilane). Polymer (Guildf) 42: 6823-6831.
- O’Brien B, Carroll W (2009) The evolution of cardiovascular stent materials and surfaces in response to clinical drivers: A review. Acta Biomater 5: 945-958.
- Derkaoui SM, Labbé A, Chevallier P, Holvoet S, Roques C, et al (2012) A new dextran-graft-polybutylmethacrylate copolymer coated on 316L metallic stents enhances endothelial cell coverage. Acta Biomater 8: 3509-15.
- Sridharan V, MeneÃŒÂndez JC (2010) Cerium(IV) Ammonium Nitrate as a Catalyst in Organic Synthesis. Chem Rev 110: 3805-3849.
- Michel EC, Montaño-Machado V, Chevallier P, Labbé-Barrère A, Letourneur D, et al (2014) Dextran grafting on PTFE surface for cardiovascular applications. Biomatter 4: e28805.
- Okieimen FE, Ogbeifun DE (1996) Graft copolymerizations of modified cellulose, grafting of acrylonitrile, and methyl methacrylate on carboxy methyl cellulose. J Appl Polym Sci 59: 981-986.
- Okieimen FE, Ogbeifun DE (1996) Graft copolymerizations of modified cellulose: Grafting of methyl acrylate, ethyl acrylate and ethyl methacrylate on carboxy methyl cellulose. Eur Polym J 32: 311-315.
- Chauvierre C, Labarre D, Couvreur P, Vauthier C (2003) Radical Emulsion Polymerization of Alkylcyanoacrylates Initiated by the Redox System Dextran−Cerium(IV) under Acidic Aqueous Conditions. Macromolecules 36: 6018-6027.
- Gao J-P, Tian R-C, Yu J-G, Duan M-L (1994) Graft copolymers of methy methacrylate onto canna starch using manganic pyrophosphate as an initiator. J Appl Polym Sci 53: 1091-1102.
- Lin OH, Kumar RN, Rozman HD, Mohd. Noor MA (2005) Grafting of sodium carboxymethylcellulose (CMC) with glycidyl methacrylate and development of UV curable coatings from CMC-g-GMA induced by cationic photoinitiators. Carbohydr Polym 59: 57-69.
- Yang F, Li G, He Y-G, Ren F-X, Wang G (2009) Synthesis, characterization, and applied properties of carboxymethyl cellulose and polyacrylamide graft copolymer. Carbohydr Polym 78: 95-99.
- Deshmukh SR, Singh RP (1987) Drag reduction effectiveness, shear stability and biodegradation resistance of guargum-based graft copolymers. J Appl Polym Sci 33: 1963-1975.
- Ukai S, Yoshida I, Honda A, Nagai K, Kiho T (1992) The distribution of carboxymethyl groups in O-(carboxymethyl)ated (1 → 3)-β-d-glucans and (1 → 3)-α-d-glucans. Carbohydr Res 224: 201-208.
- Ceresa RJ (1961) The synthesis of block and graft copolymers of cellulose and its derivatives. Polymer (Guildf) 2: 213-219.
- Gaylord N (1972) A proposed new mechanism for catalyzed and uncatalyzed graft polymerization onto cellulose. J Polym Sci Part C Polym Symp 37: 153-172.
- Gaylord NG, Anand LC (1972) Cellulose graft copolymers. II. Grafting of methacrylonitrile in the presence of ceric ion or ammonium persulfate. J Polym Sci Part B Polym Lett 10: 285-294.
- Gaylord NG (1976) Cellulose-Monomer Interaction and a Revised Mechanism for Graft Copolymerization. J Macromol Sci Part A - Chem 10: 737-757.
- Jenkins DW, Hudson SM (2001) Review of vinyl graft copolymerization featuring recent advances toward controlled radical-based reactions and illustrated with chitin/chitosan trunk polymers. Chem Rev 101: 3245-3273.
- Cerrada ML (2005) Introduction to the Viscoelastic Response in Polymers. In: Therm. Anal. Fundam. Appl. to Mater. Charact. Universidade da Coruña, pp 167-182
- Watanabe H (1999) Viscoelasticity and dynamics of entangled polymers. Prog Polym Sci 24: 1253-1403.
- Tranchida D, Piccarolo S, Soliman M (2006) Nanoscale Mechanical Characterization of Polymers by AFM Nanoindentations: Critical Approach to the Elastic Characterization. Macromolecules 39: 4547-4556.
- Lentz CW (1964) Method of making silicone rubber fillers.
- Swanson JW, Lebeau JE (1974) The effect of implantation on the physical properties of silicone rubber. J Biomed Mater Res 8: 357-67.