Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Review Article, Int J Cardiovas Res Vol: 6 Issue: 1
Understanding Heart Failure
| José Fernando Guadalajara-Boo* | |
| National Institute of Cardiology, Ignacio Chávez, Mexico City, Mexico | |
| Corresponding Author :
José F Guadalajara-Boo National Institute of Cardiology, Ignacio Chávez, Juan Badiano Num 1, Sección XVI, Tlalpan, 14080 México, City, México Tel: (55) 5655-2924 E-mail: guadalajara@cardiologia.org.mx |
|
| Received: November 12, 2016 Accepted: December 06, 2016 Published: January 06, 2017 | |
| Citation: Guadalajara-Boo JF (2017) Understanding Heart Failure. Int J Cardiovasc Res 6:1. doi: 10.4172/2324-8602.1000296 |
Abstract
Heart failure is a disease in which there are multiple definitions and the concept is adopted arbitrarily by each author, in such a way that the terminology is so confusing. Now the authors of the mega trials in medical treatment of heart failure avoided this confusion and refererd that this disease can be treated and have set to indicate the value of the ejection fraction- <40 or 35%. The main goal of this paper is to promptly revise the concepts of contractility, ventricular function, preload, afterload myocardial oxygen consumption (MVO2), heart failure, compensation mechanisms. These definitions and concepts are based on the original contributions of recognized researchers, in an attempt to clarify the concepts, which was born in the nomination of heart failure, and thus to avoid an erroneous interpretation, always motivated by inadequate simplification of terms, in order to scientifically explain the concept of ventricular function and heart failure.
Keywords: Ventricular function; Heart failure; Compensatory mechanism
Keywords |
|
| Ventricular function; Heart failure; Compensatory mechanism | |
Introduction |
|
| In heart failure study, it has obtained transcendent achievements in knowledge of pathophysiology, as well as the therapeutic aspects with new drugs [1-8] with electrical devices to prevent sudden death, or to optimize left ventricular function [9], revascularization procedures when the cause is hibernating myocardium [10], cardiac transplantation [11] and methods of ventricular assistance [12-13], when heart failure is a terminal process. However, the physician has been overwhelmed by the explosive advent of a repository of knowledge and this has brought by consequence that applied in daily practice, has fallen frequently in the oversimplification of the concept, in others, in the misrepresentation of them and finally, in others, in the misinterpretation of Physiology and cardiovascular pathophysiology, to the extent that these interpretations are different to the real facts, lead to mistakes at the time of interpreting research studies, the clinical picture, the results of the information of the imaging or laboratory findings, as well as the indication of therapeutic procedures, interpreting results for knowledge of natural history and prognosis of these patients and this problem is extensive to in the design of guidelines, or consensus for the diagnosis and treatment of this disease. | |
| The analysis of research studies which, over time, have allowed us to know and understand the mechanisms that govern the ventricular function and its alteration by disease; In addition, also the cause of the symptoms that given place and its meaning in the pathophysiology of the disease and finally the therapeutic measures that are those that improve the patient symptoms or which reduce their mortality; When the doctor understands the disease, is directed towards a rational meditation that no move away from the scientific evidence, allows us to reach conclusions that are valid to judgments and recommendations, especially on issues that are controversial and not infrequently cause of confusion and misapplication of treatments, as well as responses inconclusive discussions in different forums or consensus to not understanding the central theme that gave rise to the discrepancy. Indeed, today this problem is so serious that Cardiovascular Research Journal [14] conducted a survey in which the “definition of heart failure” asked to 2238 expert cardiologists. 1018 responses, of which only 30 included the definition of “Heart failure” were received. Of all the versions, none offered a conceptual response that was accepted, the conclusion was “heart failure is the label for a cardiovascular syndrome that is lacking uniform criteria for definition”[14]. | |
| The conclusion that can be reached is: “If you want to know if you understand something, try to explain it, if you can’t is that you don’t understand it”. In this paper, we review basic concepts which throughout history, great investigators have given us support to know the function of the heart and heart failure, in an attempt to conceptualize them in a way that is possible to discuss the theme environment to these basic principles and to arrive at a consensus based on the scientific evidence. | |
| In the most recent European guidelines [15] for the diagnosis and treatment of Heart Failure it is clearly defined that preserved systolic function corresponds to patients with EF>50% with symptoms of diastolic dysfunction. It also stated that patients with EF between 40 and 49% belongs to a “gray area” with slightly reduced heart failure (HFmr EF), stage that is well studied by Wanget et al. [16] in 2003 showing that patients with an EF between 40 and 49% (HF mr EF) asymptomatic have a year mortality of 1%, at 5 years of 30% and 10 years of 70%, very different from the mortality of HF r EF with 2%, 40% and 82% and 1%, 3%, 4% and 7.6% in diastolic dysfunction (HF p EF) in the same periods of time [16], so it can be concluded that patients with an EF between 40 and 49%, are in a stage of HFmrEF that is significantly different to patients with HFpr EF and EF. | |
| On the other hand, despite these new guidelines, there is no concrete definition of the disease since heart failure is defined by its “typical symptoms”, with which can occur when the heart is not in heart failure (sever mitral stenosis, constrictive pericarditis, etc.), so it is not fair, define disease only by their symptoms. | |
| In this paper we review the basic concepts of heart function and dysfunction through the history. Great researchers have given us sustenance to know the function of the heart and heart failure, in an attempt to conceptualize them in a way that it is possible to discuss the issue revised around these fundamental principles and can reach a consensus based on scientific evidence. | |
Heart function |
|
| “The heart is a muscular pump that generates pressure and move volume, its function is to supply of oxygenated blood to the tissues of the body and send unsaturated blood to oxygenate the lungs to sustain life” [17,18]. | |
| The heart has a prodigious function that takes place during the life of living beings that have blood circulation; in other words, the heart is the most perfect pump that the nature has created, that it is able to have 583 million, 200 thousand contractions and relaxations in a healthy 75 year old man since their internal biological systems save energy and optimize the function. No artifact created by man, which can able to perform a function that is comparable to the heart, without fault and within the time. Only when heart function is understood in all its magnitude, then the actual meaning of “heart failure” can be understood and avoid to minimize the symptoms or the response to treatment or other fragmentary pathophysiological concepts. | |
| In the 20th century, there are hundreds of papers of basic and clinical research on left ventricular function and heart failure, made by great Scientist men. At this writing, is intended to analyse those studies that can clarify and give scientific support to achieve an understanding of the concept of heart failure. | |
| Contractility: | |
| Contractility is the intrinsic capacity of the myofibril to shorten its length independent of the pre and afterload. In The isolated myofibril contractility can be measured by quantifying degree and speed of shortening to stimulate it directly to a constant initial length and without resistance to its shortening (no-load) [19,20]. The intact heart contractility is very difficult to quantify its function and is always subjected to a diastolic load (preload) and a force that has to overcome during his emptying (afterload). | |
| Today the closest way to meet the heart’s intrinsic contractile State is the end systolic relationship stress/volume or pressure/volume at which generates a curve that is extrapolated to pressure 0, this curve has been called “End systolic elastanse” (Emax ) [21]. This implies that a ventricle has more contractility when reduce the systolic volume higher magnitude to one greater afterload than another whose systolic volume is higher for the same afterload [21], unfortunately the method is little practical and difficult to achieve in clinical settings [22], but it is very useful for basic and clinic research [23,24]. | |
| It’s common for the term “Contractility” is erroneously used interchangeably to refer the “ventricular function”: ventricular function refers to the relationship between contractility, and instantaneous hemodynamic load (preload and afterload), and do not mean intrinsic contractile State (heart contractility) [19,20]. | |
| Wall stress (laplace law) [25,26] | |
| Wall tension: is the force that tends to separate the myofibrils in length (cm) [25]. | |
| Wall Stress It is the force that tends to separate the myofibrils in area (cm2) [25]. | |
| Preload | |
| It is the length of the myofibril at rest, immediately before ventricular contraction. In the intact heart is represented by the diastolic volume than in normal conditions generates a force that increases the area of the myofibril inmediately before contraction (diastolic wall stress) [24,25]. | |
| Under normal conditions, both the preload (diastolic volume and diastolic wall stress) and cardiac output are normal. When increases the preload: increase in diastolic volume for any reason: (aortic regurgitation or mitral insufficiency) or when using the Frank Starling mechanism to offset the fall in cardiac output heart failure (Figure 1), even without an increase in heart rate (Table 1). | |
| Figure 1: In the normal ventricular function curve: cardiac index (CI) and diastolic ventricular pressure (DP) are in normal limits. In heart failure, the curve deviates downward; and with normal DP (10 mmHg) the IC is 1.7 L / m2 / that is not enough to maintain in normal limits the blood pressure and tissue perfusion (tissue hypoperfusion).By other hand, the increase of diastolic volume, increases the CI, blood pressure and tissue perfusion it restores to normal cardiac function, but appears cardiomegaly and pulmonary congestion (dyspnea). | |
| Table 1: Compensatory Mechanisms of preload. | |
| In Figure 1, it is shown that the presence of heart failure, left ventricular function curve shifts downward so for a normal diastolic volume, cardiac output would be reduced insomuch that it would be impossible to maintain an adequate, cardiac output and tissue perfusion pressure: cardiogenic shock. The use of Starling mechanism allows normalizing cardiac output at the expense of increased diastolic volume (cardiomegaly) and therefore, diastolic pressure in the left ventricle (pulmonary congestion) and its limit is pulmonary edema. | |
| It must be emphasized that the increase in preload (heart failure) implies increased diastolic wall stress (stretch per cm2 of the myofibril), for which not all the increase in diastolic pressure implies increasing preload; as well, which brings as a consequence increased diastolic pressure. In stiff ventricles (restrictive cardiomyopathy) left ventricular diastolic pressure, increases without increasing diastolic volume, and preload remains normal (Figure 2). | |
| Figure 2: The left ventricular wall stiffness of the causes increased diastolic pressure without increasing diastolic volume. In heart failure the increase of LV pressure it is due to increase of ventricular volume and myocardial wall stress (preload) [68]. | |
| This is why, to interpret correctly the hemodynamics Forrester classification in acute myocardial infarction in four subsets [27] patients who are in subset 2 (normal cardiac output and high Wedge pressure) not necessarily is in heart failure and to ensure the diagnosis will need an Echocardiogram: dilated left ventricle and low EF is low, the diagnosis is heart failure (mortality 26–30%) (Figure 3B) [27,28] and conversely, if the left ventricle has normal dimensions and normal EF, the diagnosis is diastolic dysfunction (5% mortality) (Figure 3A) [28]. | |
| Figure 3: Myocardial infarction and pulmonary edema. | |
| Acute Diastolic Dysfunction. Heart size is normal and concur with pulmonary edema. | |
| Echo: The diastolic volume and EF are normal (normal systolic function). The heart is not using Starling mechanism. Mortality 1 - 5% [27,28]. | |
| Acute Heart Failure: Increased of heart size (Cardiac index 0.64) and pulmonary edema. The heart is using Starling Mechanism. | |
| Echo: The diastolic volume increased and EF decreased (28%). Mortality 18 – 23% [27,28]. | |
| Afterload | |
| “is defined as the force per unit sector area that opposes the ventricular contraction during the emptying of the heart towards the great vessels and obeys to Laplace law, so it is quantified by calculating the systolic wall stress [25,26]. | |
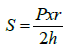 |
|
| During the isovolumic contraction intraventricular pressure increases rapidly, therefore due to wall stress; the aortic valve opens, myocardial contraction has to beat wall stress for myofibrils shortening (afterload) [18,25,26], higher pressure or intracavitary radio: greater systolic wall stress (higher afterload), and vice versa; to greater systolic thickening and lesser radio during systole: less systolic wall stress (less afterload) [18,26]. The importance of the thorough understanding of this concept, is that it is not uncommon to erroneously to refer the aortic systolic pressure as equivalent to afterload as the sole determinant, which is inaccurate. | |
| Maximum systolic wall stress is one of the main determinants for to produce myocardial hypertrophy [29]. | |
| and the mean systolic wall stress, is one of the main determinants for to increase miocardial oxygen consumption (MVO2) [30,31]. | |
| Systolic left ventricular function | |
| “Left ventricular function is the result of the simultaneous interaction of contractility with load (preload or afterload) and is quantified by ejection fraction (EF)” [32,33]. | |
| It is worth mentioning that the normal values of EF: 67 ± 8% [ 32,33]. “Diastolic heart failure” was initially defined when appeared symptoms such as Dyspnea or edema and EF was greater than 50% [34]. However, in some papers, is cataloged in an arbitrary form as “Diastolic heart failure” when the EF is between 40 and 49% [35], and in other papers classified when the EF is greater than 30% [36];when actually, these patients present heart failure with reduced systolic function, in these studies, the heart also is dilated (increased diastolic diameter), by increase pre-load (Figure 1). In those patients are erroneously classified as “Diastolic heart failure” with the implications for prognosis, treatment and mortality, since the EF is the biggest predictor of mortality [16,28];mortality is significantly higher in heart failure than “diastolic heart failure” [37]. | |
| When ventricular function and hemodynamic loads (pre-and afterload) are normal left ventricular function and MVO2 also remain in normal values [30,31]. The increase in preload increases cardiac output (Starling mechanism) [24], even when left ventricular function is depressed, works as a compensatory mechanism in heart failure (Figure 1) (Table 1). | |
| On the other hand, the increasing of the afterload means greater resistance to ventricular emptying and therefore reduces the efficiency of ventricular function; in other words, left ventricular function has an inverse relationship with the afterload, which has been shown both in experimental [21] as in clinical practice [26,38]. The greater the afterload more the depression of ventricular function (EF). | |
| Ventricular function and contractility | |
| Respect to the concepts of contractility and left ventricular function, is that quite often, both terms are used as synonyms, but they are not; so, normally the ventricular function depends on contractility: contractility Normal = Normal Ventricular function. Decreased contractility=heart failure. However it is not always so, because the Preload and afterload influence importantly Ventricular function when they are altered; in fact, an excessive increase of diastolic volume (preload), as in huge aortic, or mitral regurgitation; also patent ductus arteriosus (PDA) in newborn may cause heart failure, without intrinsic myocardial damage. The heart failure disappears when the appropriate surgical treatment corrects the overload [23,39]. | |
| In addition, the excessive increase of the afterload (as it may happen in critical aortic stenosis, coarctation of the aorta of the newborn or in hypertensive crisis), appears congestive heart failure that is refractory to any treatment and it produce death of the patient, if it is not relieved by reducing the afterload with medical or surgical treatment (aortic prosthesis valve, blood pressure reduction or surgical treatment of coarctation) in other words, in these cases, surgical treatment or rapid reduction of blood pressure are saving measures [40]. Conversely, in significant mitral regurgitation, during systole an important amount of blood return to left atrium which is a low pressure area, and left ventricle increases EF because afterload is low [41], this allows an increased systolic thickening and therefore afterload remains low. In these cases excessive increase in diastolic stress without a proper interstitial collagen support, over time can produce myocardial damage [42] then gradually appears contractile failure, reduce EF to normal Values. (Normal ventricular function with intrinsic contractile failure), at this time, the afterload is normalized and EF can be normal [41,43] and then myocardial damage is “masked”. | |
| When dissociate ventricular function and contractility: (excessive afterload and decreased EF), or otherwise, depressed contractility with normal EF, John Ross Jr. Labeled as Afterload mismatch [43]. (Table 3). | |
| Cardiac Reserve | |
| Cardiac Reserve is the ability of the heart to increase cardiac output [44]. | |
| A) Chronotropic reserve: is the ability of the heart to increase the cardiac output by increasing heart rate [43]. | |
| B) Diastolic reserve: It is the ability of the heart to increase cardiac output through the Frank-Starling mechanism, (Figure 1) and its limit is pulmonary edema”[17,44]. | |
| C) Systolic reserve: “is the ability of the heart to increase cardiac output through increasing its contractility, which depends on the anatomofunctional integrity of the myofibril [44,45] (shifts upward Starling curve) [24,47]. | |
Heart failure |
|
| In 1967, Braunwald et al. [46] defined heart failure: “When the heart loses its ability to supply enough blood to meet the metabolic needs of the tissues of the body in a normal physical activity”. | |
| This definition conceptualized with clarity that heart function ceases, it is not able to fulfil its vital role, i.e. the tissue perfusion to sustain life and that is why, if it is not corrected in a period of hours or days, ensues death.This condition in 1970, Mason et al. [45], called “Decompensated Heart Failure” and they noted that when this picture appears, the organism avoid this lethal condition, using mechanisms that attempt to restore cardiac output and tissue perfusion; these mechanisms they call “Compensatory mechanisms”; who are those are trying to restore the heart vital function: the tissue perfusion. Table 2 lists the fundamental HF compensation mechanisms [44-47]. | |
| Table 2: Compensating Mechanims in Heart Failure. | |
| Table 3: Contractility – Ventricular function relationship. | |
| The Frank Starling mechanism increases ventricular diastolic volume and normalises cardiac output (Figure 1) (Table 1). The enlargement of heart (cardiomegaly), and increased diastolic pressure with increase left ventricular wall stress, triggers secretion of brain natriuretic peptide (BNP) [48]; by other hand, the increase atrial wall stretching (stress) triggers ANP secretion [49] (Figure 4). These substances really are hormones that function as internal diuretics, and allow relieve the congestion pulmonary and systemic venous, which gets a hemodynamic State, that while the heart is in failure (decreased EF), the patient maintains cardiac output and therefore tissue perfusion (Compensated Heart Failure) and absence of systemic and pulmonary congestion and this state, allow that the patient is in functional class I, so in these conditions the patient has Asymptomatic Compensated Heart Failure (Figure 4). In these cases ACE Inhibitors have proven to be the most effective treatment to prevent the progression to symptomatic heart failure [6]. Shah PK et al. [52] in 1986 studied the ventricular function in patients with heart failure in acute myocardial infarction and they found that a 51% patients who were in functional class I (asymptomatic), had EF below 50%, 19% had EF 30% or lower (Figure 5); which denotes that the Starling mechanism (Figure 4B) and natriuretic peptides (Figure 4A) keep this hemodynamic state [24,44,45]. | |
| Figure 4: Heart Failure in functional class I. Starling mechanism maintains a normal cardiac output (CO) and natriuretic peptides induce diuresis and prevent pulmonary congestion. The patients with heart failure are asymptomatic (functional class I). |
|
| Figure 5: EF in Acute Myocardial Infarction and functional class. Note the number of asymptomatic patients with reduced EF, and in functional class I |
|
| When heart failure appears and the Starling mechanism is not able for maintain cardiac output, stimulates adrenergic system and catecholamine secretion increases the heart rate (chronotropic reserve) [44], and the positive inotropic effect increases contractility, shifts upward the Starling curve (inotropic reserve) [44,45], increasing cardiac output; and also stimulates RNA system (RAA) [1,2,53,54], and these mechanism increase cardiac output and tissue perfusion. By other hand, the secretion of angiotensin II increase peripheral resistance and maintain the perfusion pressure; aldosterone secretion retains renal Na+ and water increases the intravascular volume, the preload and cardiac output and compensate heart failure but produce clinical consequences: tachycardia, pallor, oliguria, increases of heart size, dyspnea, edema, pulmonary congestion and hepatomegaly. In conclusion, the patient’s symptoms are not by themselves due to heart failure, they are consequence due to activation of the compensatory mechanisms, but are those who maintain tissue perfusion and life, and then this condition corresponds to symptomatic compensate Heart Failure (Figure 6B). This compensation does not refer to the patients has symptoms, it refers to life preservation. | |
| Figure 6: Compensated Heart Failure (Functional Class I) Compensated asymptomatic (Functional Class III) Compensated symptomatic |
|
| Figure 7B discusses the mechanism of Frank Starling, as compensation in heart failure process; in the patient with heart failure shifts downward Starling curve (contractile failure) can normalize cardiac index and thereby restore and tissue perfusion (Compensated Heart Failure), even though is cause of increase heart size and pulmonary congestion (shortness of breath). Furthermore, when this mechanism fails to restore cardiac output appears tissue hypoperfusion (the heart loses its vital functions), which corresponds to cardiogenic shock [59] (decompensated heart failure). | |
| Figure 7: Ventricular Function Curve in Normal Heart (A), in Compensated Heart Failure (B) and in Decompensated Heart Failure (C). A) LVEP and CI in healty people. B) Compensated HF: The increase of LEDV and pressure, normalizes CI and tissue perfusion. C) Descompensated HF: when heart is unable to support the aortic perfusion pressure despite using compensatory mechanisms, appears Cardiogenics Shock. | |
| Decompensated Heart failure | |
| It is the inability of the heart to eject one sufficient amount of blood to maintain an adequate blood pressure to perfuse oxygen to the tissues of the body. This inability is due to ineffective contraction myocardial either by intrinsic damage of the myofibril or excessive hemodynamic overload [45,46,55]. | |
| When contractility is depressed in potentially reversible (hibernating myocardium) systolic reserve is lost until the cause (pharmacological, surgical or Interventional coronary reperfusion) is solved and restores tissue perfusion and life is preserved [10,56]; but when there is extensive destruction of myofibrils by necrosis (infarction) or inflammation (myocarditis), as in cardiogenic shock [25], systolic reserve is lost and the application of inotropics is not follow of improvement of EF [55]; so it does not increase the cardiac output by this mechanism (loss of the systolic reserve) and appears | |
| Decompensated Heart Failure: | |
| Cardiogenic shock [25,27], this concept, explains the reason for the reduction of mortality of cardiogenic shock with early reperfusion, retrieving the viable myocardial not functioning (hibernating myocardium) at risk of necrosis [10,56] and restores the systolic reserve, the cardiac output and life of the patient. | |
| When there is extensive myocardial damage and extreme downward deviation of ventricular function curve occurs irreversible cardiogenic shock [27,55,56], without effective treatment, leads to death (Figure 7). The clinical manifestations are: weak pulse, blood systolic pressure<80mmHg, peripheral vasoconstriction, cold, wet and bluish skin, oliguria (<50cc/hr), mental confusion, metabolic acidosis, are the true symptoms of Decompensated Heart Failure (Figure 7) in other words the inability of the heart to maintain its vital function (tisular perfussion) [27,46,55]. | |
| Myocardial hypertrophy | |
| The myocardial hypertrophy is an Adaptive mechanism that maintains normal diastolic wall stress (preload) in volume overload (aortic insufficiency) [57,58], and systolic wall stress (afterload) in systolic overload [59,60] to normal ventricular function maintenance (EF) and also MVO2 [30,31] (adequate hypertrophy) [61,62]; when the hypertrophy is unable to maintain normal diastolic wall stress in volume overload or maintain normal systolic wall stress in pressure overload, becomes inadequate hypertrophy [61,62] and appears heart failure, then the Frank Starling mechanism, adrenergic activation and RNA system appears as compensatory mechanisms; in this way the patient falls in Symptomatic Compensated Heart Failure [18,44,45]. | |
| When we understand the pathophysiologic mechanisms that operate and perpetuate the clinical Picture of heart failure, we can reach the definition of this disease: | |
| Heart failure | |
| “It is a condition in which functional or structural diffuse damage of the myofibril (Hibernation, necrosis, apoptosis or inflammation), or an excessive hemodynamic overload, causes decrease in contractile force in the heart (hence the EF); with increase the ventricular volumes with (decompensated) or without (compensated) reduction of cardiac output”[18,22,45]. | |
| In conclusion; the patient may have heart failure functional class I, by the action of the mechanisms of Starling and natriuretic peptides [45,48,49] (Figure 4), Asymptomatic Compensated Heart Failure. When Starling mechanism is unable to maintain a normal cardiac output, other compensation mechanisms (adrenergic activation and RNA) [53,54] normalizes cardiac output, but leads to symptoms that can be progressive (functional class II, III or IV) and gradually get to incapacitate the patient, but thanks to them is to restore cardiac output, tissue perfusion and life Symptomatic Heart Failure Compensated [44,55]. | |
| When the compensatory mechanism are unable to mantain cardiac output and tissular perfussion appears Decompensated Heart Failure. (Cardiogenic Shock) [27,55]. | |
| Progression | |
| When compensated heart failure is perpetuated in time, the stimulation of the compensatory mechanisms sustained in time, have deleterious effects both in the biology of myocardial function and cardiovascular hemodynamics by themselves promotes the progression of heart failure to the clinical deterioration and finally to death [1,2,63]. It is worth mentioning that the activation of the neuroendocrine system that characterizes patients with heart failure does not happen in patients with heart failure and “preserved systolic function” [64]. | |
| The understanding of the pathophysiology of heart failure [1,2], has allowed scientists develop drugs that block the deleterious effect of sustained activation of the adrenergic system (beta blockers [3], and digital, [4,5,65], the Renin-angiotensin-aldosterone system [6,7], and the aldosterone [8], have been the most effective measures to reduce mortality of these patients. On the other hand, ventricular assist systems [12,13], three chamber pacemakers and defibrillators [9] coronary revascularization in patients with myocardial viability[10] and eventually heart transplant [11], which is what really has helped even more to prolong life of patients with heart failure. It is important to emphasize that the studies that have been shown extend life in patients with heart failure, have no effect in patients with EF>50% [66]. | |
| Despite the great advances in the medical therapy [3-8,50], intervencionist [56], or surgical [10,11], mortality of heart failure is still high, and the biggest factor that influences the prognosis is the EF [16,28]. | |
| Patients who have symptoms like shortness of breath and EF>50% (heart failure “with preserved systolic function”) have a mortality rate of 7.6% at 10 years [37], while patients who have heart failure in functional class I (40-50% FE) have a mortality rate of 70%, 82% (FE<40%) and 90% in patients functional class II to IV to 10 years; when the ejection fraction is <50% [16], and the difference in mortality between two groups is explained because patients with heart failure and “preserved systolic function” do not activate the neuroendrocrine system [64], therefore only a small percentage of them presented progression toward death [67]. | |
| It is noteworthy that diastolic dysfunction (HFpEF) tend to occur in older people and mortality is higher due to co-morbidities inherent in his old age: cancer, obstructive lung disease, dementia, EVC and only 11% in men and 15% of women die from progression of heart failure, contrary to what happens in HFrEF in which progression is death bed in about 50% of cases [67]. | |
Conclusion |
|
| When the basic and clinical research studies which were already published are carefully analyzed, they offer us the scientific evidence with which it is possible to understand the meaning of heart failure, also the mechanisms that govern the pathophysiology, clinical picture and progression of this disease, allows us to clearly identify it for certain therapeutic measures applied and differentiate it from other entities that share some of their symptoms which is a cause of confusion, but that do not correspond to the disease called heart failure. | |
References |
|
|
|