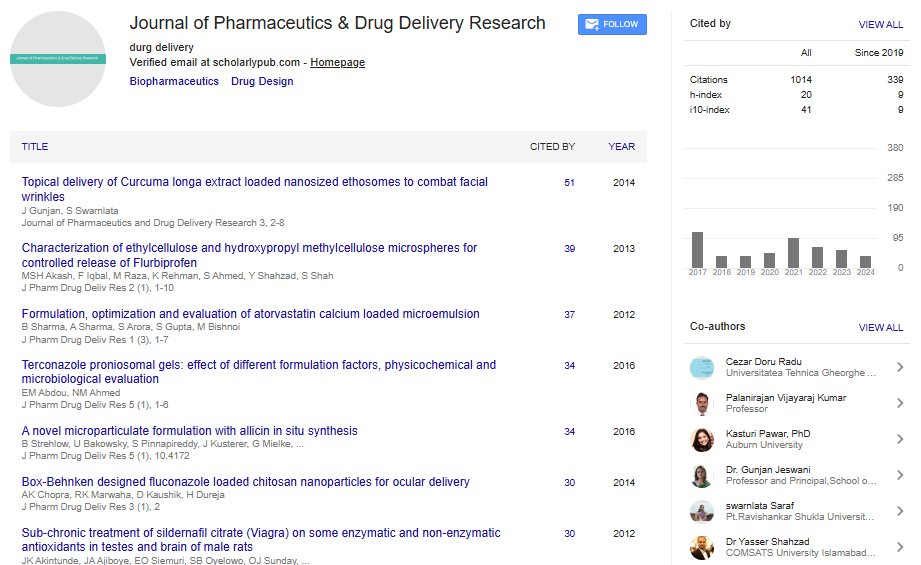
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 2 Issue: 2
Block Copolymer Crosslinked Nanoassemblies Co-entrapping Hydrophobic Drugs and Lipophilic Polymer Additives
| Daniel Scott and Younsoo Bae* |
| 1Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone, Lexington, KY 40536, USA |
| Corresponding author : Younsoo Bae Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone, Lexington, KY 40536-0596, USA Tel: +1-859-323-6649; Fax: +1-859-257-7564 E-mail: younsoo.bae@uky.edu |
| Received:: March 26, 2013 Accepted: May 21, 2013 Published: May 23, 2013 |
| Citation: Scott D, Bae Y (2013) Block Copolymer Crosslinked Nanoassemblies Co-entrapping Hydrophobic Drugs and Lipophilic Polymer Additives. J Pharm Drug Deliv Res 2:2. doi:10.4172/2325-9604.1000116 |
Abstract
Block Copolymer Crosslinked Nanoassemblies Co-entrapping Hydrophobic Drugs and Lipophilic Polymer Additives
Block copolymer crosslinked nanoassemblies (CNAs) were synthesized to investigate their patterns of entrapping and releasing an anticancer drug (17-AAG) in the presence of a lipophilic polymer additive (polylactic acid: PLA) in the core. The CNAs were prepared by crosslinking poly (ethylene glycol)-poly (aspartate hydrazide) (PEG-Hyd) block copolymers through an amide bond. The CNAslocked in the particle size after co-entrapping 17-AAG and PLA (< 50 nm in diameter). PLA with various molecular weights (3.4, 6, and 7.5 kDa) was used as polymer additives that are small enough to diffuse into the CNA core.
Keywords: Nanoparticles; Nanoassemblies; Crosslinking; Drug delivery;Controlled release
Keywords |
|
| Nanoparticles; Nanoassemblies; Crosslinking; Drug delivery; Controlled release | |
Introduction |
|
| Controlling the location and timing of drug action in the body is important for successful treatments of various human diseases [1,2]. One example is cancer chemotherapy that is often accompanied with various issues such as low therapeutic efficacy, high toxicity, and drug resistance, primarily due to the lack of an efficient and safe method to deliver drug selectively to tumors [3,4]. In this regard, nanoscale particles have been developed as carriers for controlled delivery of anticancer drugs in the body [5-7]. Nanoparticles are small enough to go through leaky tumor blood vessels, yet too large to get eliminated from the body through renal clearance [8,9]. As a result, nanoparticles increase concentrations of drug payloads selectively in tumors and spare normal tissues [10,11]. | |
| Nanoparticles can be prepared by using molecular assemblies from self-assembling or crosslinked block copolymers [12-14]. In our previous studies, we demonstrated that block copolymer crosslinked nanoassemblies (CNAs) would be viable drug carrier platforms with improved particle stability and biocompatibility [15,16]. The CNAs have a hydrophilic shell and functional core, which can be used to prevent protein adsorption in the body and to entrap various payloads for therapy and imaging [17,18]. The CNAs showed greater reproducibility in producing particles with a uniform shape and size in comparison to other existing drug carriers used in several clinical trials (e.g. polymer micelles) [15]. However, the CNAs tend to release drugs that are loaded through physical entrapment relatively quickly in comparison to the CNAs entrapping drugs through chemical conjugation. We hypothesized that if CNAs concurrently entrap a hydrophobic drug and lipophilic polymer additive, they would slow down the drug release as a function of molecular weight of the polymer additive [19]. This study is focused on testing this hypothesis, which would lead to the rational design of other nanoparticle drug carriers that can entrap and release hydrophobic drugs in a controlled manner. | |
| Figure 1 shows our overall approach to prepare nanoassemblies entrapping an anticancer drug and lipophilic polymer additives. We prepared the CNAs by using poly (ethylene glycol)-poly (aspartate hydrazide) (PEG-Hyd) block copolymers comprising hydrophilic PEG and functional Hyd segments [20-22]. PEG provided a hydrophilic and biocompatible shell to the CNA particle, and Hyd formed a crosslinked core entrapping a model anticancer drug (17-N-Allylamino-17-demethoxygeldanamycin: 17-AAG) and polymer additives (polylactic acid: PLA). 17-AAG is an anticancer drug that inhibits heat shock protein 90 (HSP90) and more than 300 client proteins of the HSP90 (most oncoproteins) in cancer cells, but 17-AAG has poor water solubility and high toxicity in patients [23-25]. PLA is FDA-approved thermosensitive lipophilic polymer that has different glass transition temperature (Tg) as a function of molecular weight, and has been widely used in various biomedical applications [26-28]. We used three types of PLA with the molecular weight of 3.4, 6, and 7.5 kDa (Tg=26, 37, and 47°C, respectively) to adjust lipophilicity of the CNA core. The CNAs entrapping 17- AAG and PLA were characterized to determine particle size, drug entrapment, and drug release patterns at the physiological condition (pH 7.4, 37°C). | |
| Figure 1: Synthesis of block copolymer crosslinked nanoassemblies coentrapping hydrophobic drugs (17-AAG) and polymer additives (PLA). | |
| These materials were used to determine particle properties such as size, drug entrapment, and drug release patterns. Therefore, results from this study will provide a better understanding of interactions between polymer nanoassemblies with a crosslinked core and a hydrophobic drug payload. | |
Materials and Methods |
|
| Materials | |
| The following chemicals were purchased from Sigma- Aldrich (USA): adipic acid, L-aspartic acid β-benzyl ester (BLA), triphosgene, anhydrous hydrazine, N,N’-diisopropylcarbodiimide (DIC), N-hydroxysuccinimide (NHS), 4-dimethylaminopyridine (DMAP), anhydrous tetrahydrofuran (THF), anhydrous hexane, anhydrous ethyl ether, anhydrous dimethylsulfoxide (DMSO), DMSO (molecular biology grade, ≥ 99.9%), dimethylsulfoxide-d6 (DMSO-d6), phosphate buffer solution, and sodium hydroxide (NaOH). α-Methoxy-ω-amino poly(ethylene glycol) (PEG, 5 kDa) was purchased from NOF Corporation (Japan). Regenerated cellulose dialysis bags with molecular weight cut off (MWCO, 6-8 kDa) and Slide-A-Lyzer G2 dialysis cassettes (MWCO, 10 kDa) were purchased from Fisher Scientific (USA). PLA (3.4, 6, and 7.5 kDa) was purchased from Polymer Source (Canada). 17-AAG was purchased from LC laboratories (USA). | |
| Block copolymer synthesis | |
| Block copolymers were synthesized as reported previously [20-22]. Briefly, β-benzyl-L-aspartate N-carboxy anhydride (BLA-NCA) was synthesizedas a monomer by reacting β-benzyl-L-aspartate (BLA) with triphosgene (1.3 eq) in dry THF (50 mg/mL) at 50°C. BLA-NCA was recrystallized at -20°C in anhydrous hexane. PEG with an amino end group was used to initiate ring-opening polymerization of the monomers in anhydrous DMSO at 50 mg/mL under nitrogen atmosphere at 45°C. The polymerization was conducted for 2 days to obtain PEG-(β-benzyl L-aspartate) block copolymer (PEGPBLA). PEG-PBLA was collected by precipitation in ethyl ether and freeze drying from benzene. The composition of the polymer was determined at this stage by proton nuclear magnetic resonance (1H-NMR) analysis, comparing the peak areas between PEG (3.5 ppm) and benzyl groups (7.4 ppm). PEG-PBLA (100 mg/mL) was then reacted with hydrazine (20 fold equivalent to benzyl groups determined by 1H-NMR) in DMSO at 40°C for 1 h. The reaction produced PEG-Hyd, which was collected by ether precipitation and freeze drying from benzene. | |
| Synthesis of cross linked nano assemblies (CNAs) | |
| Crosslinked nanoassemblies were created by crosslinking PEGHydblock copolymer chains with adipic acid in the presence of DIC, NHS, and DMAP at 4, 4, and 0.2 molar ratios between the hydrazide groups and the carboxyl groups on adipic acid. The crosslinking reaction was conducted in DMSO at 50 mg/mL at 37°C for 72 h under gentle stirring.The reaction solution was dialyzed against DMSO, followed by 50% DMSO, and finally deionized water. CNAs were collected by freeze drying. Gel permeation chromatography (GPC) and 1H-NMR were used to confirm the synthesis of CNAs. | |
| Entrapment of drug and polymer additives to CNAs | |
| In this study, we prepared four types of CNAs, which include CNAs entrapping 1) 17-AAG alone, 2) 17-AAG and 3.4 kDa PLA, 3) 17-AAG and 6 kDa PLA, and 4) 17-AAG and 7.5 kDa PLA. CNAs were dissolved in DMSO at 50 ~ 100 mg/mL, and mixed with 17- AAG and PLA at 60°C by adjusting the mass ratio (CNAs: 17-AAG: PLA=1:0.5:0.5). The mixed solution was stirred for 72 h, followed by dialysis against deionized water. Precipitates were removed by centrifuge, and the supernatant was collected for freeze drying. The samples were stored at -20°C until use. The amount of drug entrapped in each CNA was determined by measuring the maximum absorbance of 17-AAG at 337 nm in deionized water with 80% DMSO. The drug entrapment yield is expressed as a percent by weight (wt%). Entrapment of PLA was confirmed by 1H-NMR, comparing the peaks of PEG (3.5 ppm) and PLA (5.2 ppm). Particle sizes of CNAs were determined by dynamic light scattering (DLS) measurements on a Zeta sizer Nano-ZS equipped with a He-Ne laser light source and 173° angle scattered light collection configuration (Malvern, UK). | |
| Drug release experiments | |
| Drug release patterns of CNAs were determined by a dialysis method using dialysis cassettes (Slide-A-Lyzer G2, MWCO 10 kDa, Thermo Scientific, USA). Dialysis cassettes containing 1.5 mL samples were placed in 4 L of a phosphate buffer solution (10 mM, pH 7.4) at 37°C. An aliquot of 150 μL was taken out of each dialysis cassette at the time intervals of 0, 0.5, 1, 3, 6, 24, 48, and 72 h. The aliquots taken at each time point were analyzed by measuring drug absorbance at 337 nm. All experiments were performed in triplicate, and data were expressed as mean ± standard deviation (SD). | |
Results and Discussion |
|
| Synthesis of block copolymers and CNAs | |
| 1H-NMR analysis confirmed that PEG-Hyd consisted of 5 kDa PEG and 40 Hyd repeating units (data not shown). The crosslinking of PEG-Hyd was confirmed by GPC. The molecular weight of PEG-Hyd was 10.64 kDa, which increased to ~ 137 kDa after the crosslinking reaction. Such a change in molecular weight suggests that approximately 12 ~ 13 block copolymer chains form a single CNA particle. These results were consistent with our previous observations. The particle size of the empty CNA was 26 ± 4 nm. CNAs dissolved aqueous solutions, such as deionized water, buffer solutions, and saline, and no precipitation was observed. CNAs were compatible with filter sterilization and freeze drying, which are important factors for the storage of pharmaceutical ingredients. | |
| Polymer nanoparticles, including liposomes, polymer micelles, and dendrimers, have drawn attention in recent explorations of drug delivery technology [29,30]. Polymer nanoparticles have shown great promise to protect drug payloads in in vivo environments and maintain drug concentrations for a longer term compared to free drug [12]. Nanoscale materials (< 100 nm in diameter) increase drug concentrations selectively in tumors that have leakier blood vessels as opposed to normal tissues. Despite the potential, nanoparticle drug carriers often show variable properties such as particle size, shape, biocompatibility, and drug entrapment/release yields, which change the pharmacokinetics, tissues distribution, and eventually therapeutic efficacy of the drug payload [31]. To avoid these problems, we have recently developed stable and uniform drug carriers from block copolymer crosslinked nanoassemblies, denoted as CNAs, and demonstrated that the CNAs can be used for entrapping various payloads through either physical entrapment or chemical conjugation [15,16,32]. The CNAs with physical drug entrapment, however, appeared to release drug quickly. We hypothesized that the core environment of the CNAs would play an important role in modulating the release of drug molecules from the particle. In our pilot experiments, we used various aliphatic compounds with 2 ~ 8 methylene groups to make the CNA core more hydrophobic and increase drug entrapment yields, but the particles became too large (> 200 nm) and the drug loading was less than 1% (data not shown). In this study, we attempted to modify the CNA core with flexible yet lipophilic PLA polymer and evaluated the drug entrapment and release patterns. | |
| Entrapment of drug and polymer additives to CNAs | |
| CNAs successfully entrapped either 17-AAG alone or 17-AAG and PLA in combination (Figure 2). Drug-loaded CNAs were watersoluble and readily separable by centrifuge from unloaded 17-AAG and PLA that precipitated in aqueous solutions. The concentration of 17-AAG in the CNAs was determined by measuring absorption at 337 nm. PLA entrapped in the CNAs was confirmed by 1H-NMR (Figure 3). A drug entrapment yield was 27.35 wt% when CNAs entrapped 17-AAG alone. CNAs with a combination of 17-AAG and PLA showed a lower drug entrapment yield. CNAs with 3.4, 6, and 7.5 kDa PLA entrapped 4.7, 16.6, and 12.4 wt% 17-AAG, respectively. The particle sizes of the CNAs co-entrapping drug and polymer additives were 39.7 ± 9, 49.1 ± 12, and 35.1 ± 2 nm for the 3.4, 6, and 7.5 kDa PLA, respectively (Table 1). | |
| Figure 2: Drug entrapment yield determination. | |
| Figure 3: Proton nuclear magnetic resonance (1H-NMR) spectra in DMSO-d6 | |
| Table 1: Particle characterization data summary. | |
| CNAs from PEG-Hyd offered a drug carrier with increased particle stability compared to other surfactant-based or self-assembling drug carrier formulations [17]. The CNAs, however, still released drug quickly when drugs are loaded through a physical entrapment process. Such a quick drug release pattern suggested that the CNAs would have a rigid core that can minimize the interactions between the particle and the drug entrapped, or the block copolymers might be loosely packed in CNAs. 1H-NMR partially answers this question. It is generally known that block copolymer segments that tightly packed in a nanoparticle show no 1H-NMR signal due to the lack of molecular mobility [33,34]. Other drug carriers from self-assembling block copolymers, such as polymer micelles, often show the NMR peaks only from water-soluble segments in D2O (for example, PEG) while the peaks from the core of the particle disappear due to tight packing of molecules. Whereas, the NMR spectra clearly showed the peaks of the core-forming segment of the block copolymer from the CNAs in D2O, indicating that the crosslinked core of CNA is loosely packed although the particle size of the CNA was relatively small (< 50 nm). The NMR analysis also indicates that the conformation of a polymer chain is locked the CNA core. Relatively consistent particle sizes of CNAs (Table 1) suggest that 17-AAG and PLA with varying molecular weights are present in the network of loosely packed block copolymer chains in the CNAs while the crosslinked core prevents the CNAs from growing in size after entrapping the drug or polymer additives. These results reveal that the crosslinking of block copolymers is useful to produce a nanoparticle that has a rigid core with a fixed conformation, yet it accelerates drug release from the CNAs. | |
| Drug release patterns | |
| CNAs entrapping 17-AAG alone released more than 90% of total drug entrapped within the first 3 h (Figure 4). CNAs with 17-AAG alone achieved complete drug release in 24 h. CNAs co-entrapping 17-AAG and PLA slowed down the drug release significantly, and 40 ~ 50% of drug remained in the CNAs after 3 h. PLA changed the drug release pattern according to the molecular weight. CNAs with 3.4 kDa PLA showed a plateau for about 30 minutes before they released the drug. Burst drug release was seen in CNAs with 6 kDa PLA for the first 1 h. PLA with the highest molecular weight (7.5 kDa) suppressed the burst release and removed the initial delay in releasing drug. However, the overall drug release pattern for the CNAs co-entrapping 17-AAG and PLA became similar after 3 h. CNAs with a combination of 17-AAG and PLA showed sustained drug release up to 3 days. | |
| Figure 4: Drug release from various CNA formulations (pH 7.4, 37°C). | |
| The drug molecules in the CNAs may move more freely through and exit the crosslinked core compared to other drug carriers with a tighter packed core. To slow down drug release from the CNAs, we co-entrapped a drug and polymer additive. PLA used in this study was thermo sensitive polymer with a relatively small molecular weight (< 10 kDa), which was loaded into and released from the CNAs without changing the size and solubility of the particle in water. PLA was successfully entrapped in the CNAs through the reaction at an elevated temperature (60°C). PLA has potential to reduce drug release rates of the CNAs because it is a lipophilic polymer that can make the CNA core more favorable for entrapping lipophilic drugs. Tg of PLA changes as a function of the molecular weight, and the Tg is 26°C, 37°C, and 47°C for 3.4, 6, and 7.5 kDa of PLA, respectively. We hypothesized that drug release from CNAs could be controlled by entrapping PLA with different Tg (flexibility) in the CNA core. The 3.4 kDa PLA was initially expected to show no effect on drug release pattern as it should be in a liquid state at 37°C, the temperature where the drug release experiment was conducted. Similarly, we expected no drug entrapment or release for the CNAs with 7.5 kDa PLA because the core might be too hydrophobic for drug molecules to diffuse through. Interestingly, all formulations of CNAs co-entrapping 17-AAG and PLA showed successful drug entrapment and release. However, the CNAs showed lower drug entrapment compared to the CNAs with 17-AAG alone. The CNAs with 3.4 kDa PLA showed the lowest drug loading of 4.7 wt%. The 6 kDa PLA yielded the highest drug loading of 16.6 wt% and 7.5 kDa PLA showed 12.4 wt% of drug loading in the CNAs. | |
| Drug carriers must entrap drug payloads stably in the body until they accumulate in tumors. The CNAs without PLA would release the majority of drugs before they accumulate in tumors. CNAs with PLA slowed down drug release, and approximately 50% of drugs still remained in the particle. We attempted to measure the particle sizes of the CNAs co-entrapping 17-AAG and PLA at elevated temperatures (25 50°C), but no temperature-dependent change in particle size was observed. These results demonstrate that the molecular weight and Tg of PLA did not appear to modulate drug release kinetics for the CNAs. CNAs co-entrapping 17-AAG and 3.4 kDa PLA suppressed drug release for 30 minutes and released the drug rapidly. CNAs with 6 kDa PLA released 17-AAG most rapidly in the first 30 minutes, yet the overall drug release pattern remained similar to other CNA formulations with a combination of 17-AAG and PLA. These results demonstrate that the co-entrapment of an anticancer drug and PLA would be a viable option to slow down drug release from nanoparticle drug carriers with a crosslinked core. Such drug carriers are expected to suppress drug release in the blood stream and increase drug accumulation in tumors. | |
Conclusion |
|
| The crosslinking of PEG-Hyd block copolymers produced stable and uniform particles (denoted CNAs) that can be used as drug carriers for entrapping various molecules. The CNAs, smaller 50 nm in diameter, entrapped a model anticancer drug (17-AAG) alone or in combination with polymer additives (PLA with various molecular weights). CNAs entrapping 17-AAG alone showed a 27.35 wt% drug entrapment yield and released 90% of the drug entrapped in 3 h. In comparison to CNAs with 17-AAG alone, the CNAs co-entrapping 17-AAG and PLA had lower drug entrapment yields (4.7 ~ 12.4 wt%). The addition of PLA to CNAs resulted in more sustained drug release from the particles. There was no significant change in drug release from the CNAs with different PLA formulations (3.4, 6, and 7.5 kDa). Therefore, the co-entrapment of hydrophobic drugs and lipophilic polymer additives would be a viable option to modulate drug release from CNAs without altering the particle properties. | |
Acknowledgments |
|
| This research is supported by the Kentucky Lung Cancer Research Program. | |
References |
|
|
|