 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Appl Bioinforma Comput Biol Vol: 7 Issue: 4
A Computational Approach for Mutational Analysis of KRAS Snps and Toxicity Prediction of Screened Compounds of Lethal G12R KRAS SNP
Fareha Masood*, Khalida Naveed and Syeda Tatheer Fatima
Baqai Institute of Information and Technology, Baqai Medical University, Karachi, Pakistan
*Corresponding Author: Fareha Masood
Baqai Institute of Information and Technology, Baqai Medical University, Karachi, Pakistan
Tel: 021-36617235
E-mail: s.farehamasood@gmail.com
Received: November 11, 2018 Accepted: December 24, 2018 Published: December 31, 2018
Citation: Masood F, Naveed K, Fatima ST (2018) A Computational Approach for Mutational Analysis of KRAS SNPs and Toxicity Prediction of Screened Compounds of Lethal G12R KRAS SNP. J Appl Bioinforma Comput Biol 7:4. doi: 10.4172/2329-9533.1000158
Abstract
KRAS protein is vital for normal cell signaling and its single point mutations are linked with the development of numerous forms of malignancies. Despite of various reports available on SNPs of KRAS and its unresponsiveness to available drug therapies, there is an urgent need for the development of effective and potential drug target against it. In present study, a strategy is made with the combination of computational biology, cancer informatics and cheminformatics. Mutational analysis, structure based virtual screening (SBVS) and toxicity profiling of compounds have been performed for the prediction of therapeutic drug targets against KRAS detrimental mutations. Among 31 retrieved mutations from dbSNP and UniProt, 12 mutations were predicted to be lethal by PROVEAN and PolyPhen tools. The protein stability analysis was done by using I-Mutant tool, from the 12 mutations 8 mutations have destructive effect while 4 mutations have constructive effect on protein stability. 3D-structures of 12 identified mutated KRAS genes were modeled by the aid of DS-visualizer. Among the identified lethal mutations G12R was selected for SBVS, the KRAS protein was subjected as receptor while GDP as a ligand. The SBVS was performed by using idock server, among 11630 library of compounds the best conformation was obtained having lowest idock score (-11.389 Kcal/Mol). The protein and ligand interactions were examined by using Ligplot+ and toxicity profiling of top 5 compounds were performed by using Lazar and ProTox tools. This study will be helpful in the development of effective diagnostic methodologies and drug targeted therapy against G12R mutation.
Keywords: KRAS; G12R; SNPs; Virtual screening; Cancer
Introduction
Single Nucleotide Polymorphisms (SNPs) are the one of the contributor of genetic mutation in human. At least one SNP is documented among ninety three percent of complete set of human genes [1]. Hence, most of the biological variations in an individual are the result of SNP generation. A better understanding of SNPs and their link with phenotypical characteristics can be helpful in identification of etiology of various maladies and disorders. SNPs can be classified on the basis of its occurrence in the gene segments i.e., coding SNPs, noncoding SNPs and intergenic SNPs [2,3]. The alteration in amino acid caused due to SNPs have damaging effect on protein by affecting its various properties like function, structure, stability and solubility etc. [4,5]. Further, SNPs also perturb regulation at gene level by altering binding factors of replication, transcription [6] and at the conservation of cellular and tissue integrity processes [7].
The Kirsten rat sarcoma viral oncogene homolog gene (KRAS) is a Ras isoform oncogene, positioned at 12.1 site of chromosome with 12 short (p) arm with six exons. This gene encodes ~21 kDa K-Ras protein chiefly associated with regulation of different phases of cell cycle including cell division, differentiation and programmed cell death.
This protein is G-coupled protein and as a part of RAS/MAPK pathway communicates signals from cell exterior to nucleus for cell growth, development and differentiation [8]. According to the cancer genome atlas database (TCGA) KRAS gene is one of the driver mutation in pancreatic, lung and colon cancers, KRAS gene raised the activation process of tyrosine kinase and as well as it encodes for signaling protein which is responsible for cell death and cell growth [9]. Mutated KRAS gene is still in under examination to be developed as druggable target. Up until now, researches are carrying out over KRAS mutant but it still remained as an undruggable target and linked with failure of EGFR inhibitor treatment therapy for adenocarcinomas of lung and colon [10-14]. RAS genes associated with malignancy are categorized by single base missense mutations, among them ninety-eight percent are at G12, G13 and Q61 residues. Among them, G12 residue mutations are the principle mutation found in KRAS liked carcinomas. On the basis of relative incidence of mutations at these locations, there are also various types of cancers. Like the mutations predominantly linked with G12 are pancreatic ductal adenocarcinoma (PDAC), lung adenocarcinoma (LAC) and colon and rectal carcinomas (CRC) [15].
Bioinformatics has been developed as a major and significant part of various fields of biology. Bioinformatics tools and software helps in mining and extracting suitable information from huge scrambled raw biological data; investigation of expression at DNA and protein levels, simulation and modeling of molecular structures of DNA, RNA, and protein, evaluation of hereditary and genetic data and aids examine and record the biological pathways and networks that are an vital constituent of biological system [16]. There are currently numerous published scientific work unfolding the contribution of SNPs of KRAS gene with diverse range of diseases, computational analysis has also been carry out on the functional and structural consequences of SNPs in this gene. In this study, different publicly available bioinformatics tools and databases have been used for a comprehensive analysis of SNPs in KRAS gene specifically associated with cancer and whether these SNPs contribute in disease development or not was examined. Then disease associated SNPs were selected for the identification and modeling of mutant protein and these proteins were compared with the native non-mutated protein in order to identify those alterations that affect the normal protein structure and function. After that, virtual screening of library of compounds against the most prevalent cancer causing SNP modeled protein was carried out and ligand-protein interaction was analyzed in order to predict potent and potential inhibitors against the modeled protein. In last, toxicity profile of potent inhibitors was predicted. This study will predict potent drug targets which will be helpful for the scientific community; it can be further examined by advanced experimental studies.
Materials and Methods
Retrieval of dataset
The SNPs data of human KRAS gene was retrieved from database of SNP (dbSNP) [1] and UniProt database [17]. The reference SNP (rs) IDs were retrieved from dbSNPs, the database was used with subsequent parameters: Homosapiens was selected as an organism, variation class was selected as SNP and clinical significance was selected as pathogenic. The rsIDs were further analyzed by the aid of UniProt database, the KRAS gene was searched with Homo sapiens as organism. The approaches which have been used are presented in schematic format (Figure 1).

Figure 1: Flowchart of methodology.
Identification of damaging SNPs
For the analysis whether the selected SNPs from uniprot and dbSNPs are detrimental or not, the structural and functional perturbation studies were carried out by online available tools like Protein Variation Effect Analyzer (PROVEAN) [18] and Polymorphism Phenotyping (Polyphen-2) [19]. PROVEAN software was used to predict the effect on the biological function of a protein when an amino acid is mutated. For the prediction of structural and functional change due to the SNPs, bioinformatics based program PolyPhen-2 was used. This prediction is centered on features encompassing the arrangement, structure and evolutionary conservation of amino acids underlying the substitution. The score was calculated in term of difference in position-specific independent count scores (PSIC) between two structures. On the basis of PSIC score the protein damage is categorized as safe (0.00-0.90), potentially detrimental (1.0-1.50), and possibly detrimental (1.40-1.90) and highly detrimental (2.00 and more) [20].
Analyzing stability of protein
The I-Mutant tool is based on Support Vector Machine (SVM). With the aid of I-Mutant (version 3.0) [21], the protein stability alteration due to a single mutation was determined. The prediction of protein stability changes upon single point mutation from protein sequence was selected and DDG value and binary classification method was used to determine the stability of protein due to SNPs.
Molecular modeling of mutant proteins
The standalone software Discovery Studio (DS-Visualizer) [22] was used to determine the three dimensional structure of the selected proteins and then modify the instinctive residues with a transformed one in order to study its impact on structure of protein as a step towards computational drug design. The electrostatic, hydrogen and hydrophobic interactions with in 5 Angstrom region of amino acid was also monitored by using DS-Visualizer.
Virtual screening
Visualization of protein-ligand complex plays an important role in elaborating protein ligand interactions and aid in the development of novel and new drug design. To date, dozens of visualization tools already exist: VMD, PyMOL and Chimera are very well-known and highly cited [23]. Protein-ligand docking is a key computational method in designing drug discovery process. In order to dock largescale data automatically, idock provide a user-friendly publically available web platform. This tool is not only helpful in predicting the ligand binding with protein but also give information regarding how the ligand interacts with the protein [24]. For SBVS G12R mutation was selected among 12 identified lethal mutations because G12R SNP has been identified as deadly mutation and its 3D structure is available so the docking can be performed. The SBVS was performed by using idock online software, the input parameters for docking studies were PDB format of protein (PDB: 4QL3) [25]. The job was submitted by using inbuilt parameters and 11630 compounds have been identified.
Toxicity profiling of identified potent inhibitors
For the cytotoxicity profiling of the newly identified potent inhibitors online servers Lazar [26] and ProTox [27] were used. Similar to the toxicological threat evaluation, lazar generates indigenous quantitative structure activity relationship (QSAR) models for respective compound to be predicted. The foremost aim of lazar is to offer a basic tool for the forecast of multifaceted cytotoxic endpoints including carcinogenicity, long-term and short term teratogenicity [26]. ProTox is an online freely accessible tool which used to predict toxicity of respective compounds on the basis of similarity and fragment based approaches. It provides better understanding of mechanisms involved in the development of toxicity and therefore it determines better result as compared to existing QSAR based approaches [27].
Results and Discussion
According to the current release of NCBI dbSNP (build 151) it consists of 6 different organisms and one of the organism is Homo sapiens which comprises of 335,215,764 non validated rsIDs from which 61,743 rsIDs were validated. After submitting the gene of interest KRAS in dbSNP it produced 6219 rsIDs and with specific parameters it produced 31 rsIDs which were found to be associated with clinical pathogenesis in Homo sapiens (Supplementary Table 1). The 31 rsIDs were downloaded in batch format which will be used for further analysis. The selected UniProt accession was P01116, which consists all the 31 rsIDs under the heading of natural variants all the targeted rsIDs were present and linked with ClinVar [28] which is a disease focused database.
Lethal SNPs predicted by PROVEAN and PolyPhen
All reported 31 rsIDs were submitted to PROVEAN and PolyPhen servers and both the tools predicted 10 rsIDs (rs193929331, rs104894361, rs104894365, rs104894366, rs104894364, rs121913528, rs104894359, rs121913527, rs387907206, rs121913530) to be lethal (Table 1). The mutation in proline at 34th position with arginine and leucine has been associated with rsID (rs104894366) and the mutation in glycine at 60th position with arginine and serine has belonged to rsID (rs104894359). PROVEAN tool predicted that all 12 mutations have been identified as deleterious because the scores were below to the predefined threshold (-2.5). The PolyPhen tool predicted the detrimental mutations on the basic of scores which have been discussed in methodology section, among 12 mutations A59T and G12R were possibly damaging while other 10 mutations were probably damaging.
| SNP ID | SNPs | Provean score | Polyphen result | Polyphen score |
|---|---|---|---|---|
| rs104894359 | G60R | -14.96 | Probably Damaging | 1 |
| rs104894359 | G60S | -14.96 | Probably Damaging | 1 |
| rs104894366 | P34L | -14.1 | Probably Damaging | 1 |
| rs104894366 | P34R | -14.1 | Probably Damaging | 1 |
| rs104894364 | T58I | -13.96 | Probably Damaging | 1 |
| rs387907206 | K147E | -13.57 | Probably Damaging | 1 |
| rs121913528 | A59T | -12.98 | Possibly Damaging | 0.936 |
| rs121913527 | A146T | -12.71 | Probably Damaging | 0.963 |
| rs121913530 | G12R | -11.83 | Possibly Damaging | 0.741 |
| rs104894365 | V14I | -11.31 | Probably Damaging | 0.999 |
| rs193929331 | K5E | -10.82 | Probably Damaging | 0.998 |
| rs104894361 | K5N | -10.82 | Probably Damaging | 0.999 |
Table 1: Predicted potentially lethal SNPS with PROVEAN and PolyPhen.
Protein stability change predicted using I-Mutant
The I-Mutant tool results are presented as free energy changes induced due to SNPs contributing interference to protein stability. Among the twelve lethal predicted SNPs of KRAS gene, eight SNPs V14I, P34R, G60R, G60S, A146T, K147E, A59T and G12R were found to be contributing the protein instability while the other four SNPs K5E, K5N, T58I and P34L have positive impact on protein stability. The elaborated results are presented in (Supplementary Table 2).
Modeling of amino acid substitution effects due to SNPs on protein structure
Ds visualizer program was used to visualize the possible three dimensional structures of mutant protein products of lethal KRAS gene SNPs. For this purpose, we submitted PDB format of KRAS protein (4OBE) in DS visualizer and replaced the wild-type amino acid with the mutant in accordance to the predicted lethal KRAS SNPs. This helped us in the better understanding of structural changes induced due to SNPs in native KRAS protein structure. The nature and type of amino acid in a protein is responsible for structural and functional features like hydrophobicity, rigidity, solvent accessibility, charge density and electrostatic potential etc. Due to the change of amino acid induced because of lethal SNPs in mutant KRAS gene product confer different structural and functional characteristics to the native KRAS protein. The effect of each predicted SNPs on the native protein structure is depicted as three dimensional structures (Supplementary Figures 1-12). The wild type amino acid is represented in green color; the mutated amino acid is represented in red color while the 5 Angstrom region with in the amino acid is represented in pink color.
The SNP ID (rs193929331) replaces lysine with glutamate at 5th position of KRAS protein. Both are polar amino acid, this may enhance the electrostatic interactions and hydrogen bonding that ultimately contribute to improved stability to substituted protein. The structural difference between the native and mutated protein can be seen in Supplementary Figure 1.
KRAS SNP (rs104894361) at 5th position of KRAS protein has substitution of negatively charged small chain (aspartic acid) in place of comparative large positively charged amino acid (lysine). Both are charged amino acid and involve in hydrogen bonding and salt bridge formation. The stability of mutant protein found to be increased because of aspartic acid, this may be due to small size of aspartic acid residue which may be more effective in mentioned interactions (Supplementary Figure 2).
Substitution of isoleucine instead of valine residue at position 14th of KRAS (rs104894365) depicted in comparison with wild-type (Supplementary Figure 3). Both are non-polar and hydrophobic in nature and may be buried in the core structure but isoleucine has long side chain than valine that may interrupt the overall hydrophobic interactions with adjacent residue due to bulkiness.
SNP id (rs104894366) have mutation of leucine in place of proline at 34th position, the modeled structure altered and wild-type KRAS protein are presented in Supplementary Figure 4. Proline is a cyclic non-polar secondary imino acid that reduce the structural flexibility of a protein due to rigid conformation. Both are non-polar amino acid and most probably buried in the core region of the protein. When proline is substituted with leucine non-polar hydrophobic non-cyclic amino acid causing protein more flexible that may boost overall interaction with the adjacent residue thus result in increased stability of the protein.
The mutation of proline to arginine at position 34th of KRAS (rs104894366) (Supplementary Figure 5), causes a decrease in protein structural stability. This may be due to the fact that proline provides rigidity to protein structure because of cyclic imino group and when it was switched with polar arginine involving in effective intermolecular forces.
The mutation (rs104894364) leads to a change of threonine to isoleucine at 58th residues of KRAS represented as three dimensional structures (Supplementary Figure 6). The side chain of threonine has a hydroxyl group that keenly participates in hydrophilic interactions and preferentially shields the surface of the molecule while isoleucine having hydrophobic side chain preferentially builds up the core region of the protein. As the substitution is at 58 positions and may be located in the core region therefore result in the increased stability in the mutant protein as compared to the native one.
The replacement of alanine (hydrophobic) with threonine (hydrophilic) amino acid at 59th residue located at the core region of SNP protein id (rs121913528) causes fall in protein stability because of hydrophilic and polar interaction of threonine in the core region of mutant protein (Supplementary Figure 7).
SNPs id (rs104894359) have arginine in place of glycine at 60th residues (Supplementary Figure 8). Glycine is the simplest amino acid and offer great flexibility to the protein structure as part of loop or coil. While arginine strengthens the structure with hydrogen bonding and salt bridge formation. The reduction in the stability of the mutant protein may because of lack of flexibility.
The mutation (rs104894359) is similar like mutation of (rs104894359) where glycine is replaced with polar amino acid (serine) at 60th residue of KRAS and similar outcome was observed in term of reduction in the protein stability (Supplementary Figure 9).
The SNP (rs121913527) at position 146 (core region) having threonine (hydrophilic) instead of alanine (hydrophobic) reduce the protein stability in reference to the native protein having alanine amino acid shown in Supplementary Figure 10.
The mutant protein (rs387907206) having SNP at 147 position of glutamate in place of lysine reduces the stability as compared to native KRAS protein (Supplementary Figure 11). Both are polar amino acid but lysine has comparatively long side chain as compared to glutamate providing more chance of intermolecular interaction.
SNP id (rs121913530) having arginine in place of glycine at 12th position reduced the protein stability. Glycine is the non-polar amino acid and gives flexibility to the protein and when it was replaced with polar amino acid arginine, it had been associated with salt bridge formation (Supplementary Figure. 12). According to an in vitro study G12R have an ability to be better transformed, in reported study mice with G12R mutations showed advanced stage lung tumors compared with other KRAS mutations [29].
Retrieval of therapeutic compounds from virtual screening
A total of 11630 compounds were docked after fulfilling the required criterion with the aid of idock. The results from idock server were available as hits.csv.gz (free energy, ligand efficiency, RF-Score, hydrogen bonds, molecular properties, link to substance information and supplier list of the successfully docked compounds) and hits. pdbqt.gz (conformations of the hit compounds). The top 5 compounds were selected on the basis of idock score and each compound were further investigated by submitting the SMILES to SwissADME web server for the analysis of physiochemical properties (molecular weight (MW)), lipophilicity (Log P), water solubility (log S) and drug likeness (DL) of compounds (Table 2). The DL of top 5 compounds had been determined by 5 filters which were Lipinski, Ghose, Veber, Egan and Muegge [30]. The partition coefficient of top 5 compounds was 2.99, 2.42, 2.52, 2.51 and 2.34 respectively. The gastrointestinal absorption of all the 5 compounds were high. The dock pose between the compounds and G12R protein have been presented in (Figures 2-6). For better understanding of interactions found between top 5 best predicted compounds and G12R protein, LigPlot+ [31] was used to generate the 2D Figures of protein-ligand interactions from its 3D coordinates. All the top 5 compounds displayed best possible optimal interaction with our target protein showing potential to become a lead inhibitor against G12R protein after further investigation. The schematic results of LigPlot for all these five compounds are shown in Figures 7-11.
| Zinc ID | Idock score (Kcal/Mol) |
Rf-Score (PKD) | Drug like properties |
|---|---|---|---|
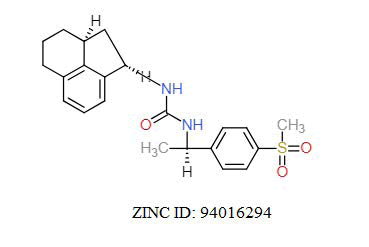 |
-11.389 | 6.902 | MW. 398.52 g/mol, LogP. 3.44, LogS. -4.33, DL. Yes |
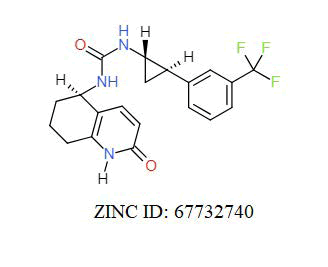 |
-11.104 | 6.826 | MW. 391.39 g/mol, LogP. 3.37, LogS. -3.65, DL. Yes |
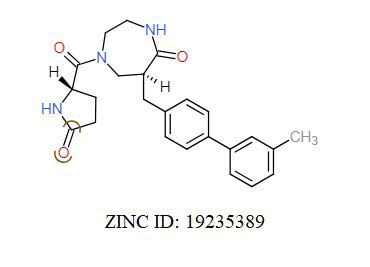 |
-10.939 | 6.707 | MW. 405.49 g/mol, LogP. 2.29, LogS -3.69, DL. Yes |
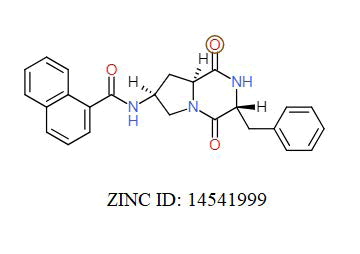 |
-10.696 | 6.827 | MW. 413.47 g/mol, LogP. 2.55, LogS. -4.55, DL. Yes |
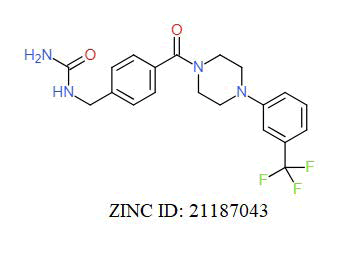 |
-10.644 | 7.221 | MW. 406.40 g/mol, LogP. 2.61, LogS -3.72, DL. Yes |
Table 2: idock results of top 5e compounds.

Figure 2: Visualization of docking of targeted protein (4QL3) with compound.

Figure 3: Visualization of docking of targeted protein (4QL3) with compound 2.

Figure 4: Visualization of docking of targeted protein (4QL3) with compound 3.

Figure 5: Visualization of docking of targeted protein (4QL3) with compound 4.

Figure 6: Visualization of docking of targeted protein (4QL3) with compound 3.

Figure 7: Ligplot showing the hydrogen bonds and hydrophobic interactions between targeted protein (4QL3) and compound 3.

Figure 8: Ligplot showing the hydrogen bonds and hydrophobic interactions between targeted protein (4QL3) and compound 2.

Figure 9: Ligplot showing the hydrogen bonds and hydrophobic interactions between targeted protein (4QL3) and compound 3.

Figure 10: Ligplot showing the hydrogen bonds and hydrophobic interactions between targeted protein (4QL3) and compound 4.

Figure 11: Ligplot showing the hydrogen bonds and hydrophobic interactions between targeted protein (4QL3) and compound 4.
Toxicity profiling analysis of top 5 compounds
The toxicity profiling of the top 5 compounds were generated by ProTox and LAZAR servers, all these compounds were found to be non-mutagenic, non-carcinogenic, non-immunotoxic and noncytotoxic except the compound 4 which was immunotoxic. The maximum recommended daily dose for human has been tabulated (Supplementary Table 3). All the compounds were non-carcinogenic that proved that the compounds could be used as possible candidates for the treatment of cancer.
Conclusion
The mutational analysis of all the identified lethal SNPs were performed, the 12 identified SNPs have damaging effects on biological functions and most of them were involved in protein instability. Virtual screening of G12R mutation was implemented because of the availability of PDB structure and G12R have been reported to be linked with various types of cancer. The virtual screening was performed with 11630 libraries of compounds following with other parameters. Among 11630 compounds, top 5 compounds were selected for toxicity profiling and they were predicted to be non-carcinogenic. The current study was implemented by using computational biology and bioinformatics strategies however, it lacks experimental evidences but bioinformatics approach has the potential to propose such therapeutic drug targets. Furthermore, experimental studies of these compounds were lead towards the development of effective drug therapy against KRAS mutation induced cancers.
References
- Chakravarti A (2001) Single nucleotide polymorphisms: To a future of genetic medicine. Nature 409: 822-823.
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, et al. (2005) The transcriptional landscape of the mammalian genome. Science 309: 1559-1563.
- Liu J, Gough J, Rost B (2006) Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet 2: e29.
- Ng PC, Henikoff S (2006) Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet 7: 61-80.
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, et al. (1994) Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. New Engl J Med 331: 1056-1061.
- Lander ES (1996) The new genomics: Gobal views of biology. Science 274: 536.
- Thomas R, McConnell R, Whittacker J, Kirkpatrick P, Bradley J, et al. (1999) Identification of mutations in the repeated part of the autosomal dominant polycystic kidney disease type 1 gene, PKD1, by long-range PCR. Am J Hum Genet 65: 39-49.
- Guerrero S, Casanova I, Farré L, Mazo A, Capellà G (2000) K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res 60: 6750-6756.
- Tomczak K, Czerwinska P, Wiznerowicz M (2015) The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol 19: A68.
- Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26: 3291-3310.
- Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3: 459-465.
- Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11-22.
- Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA (2010) Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor–targeted therapy? J Clinl Oncol 28: 4769-4777.
- Bardelli A, Siena S (2010) Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28: 1254-1261.
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014) Drugging the undruggable RAS: Mission possible? Nat Revi Drug Discov 13: 828-851.
- Stolze B, Reinhart S, Bulllinger L, Fröhling S, Scholl C (2015) Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep 5: 8535.
- Consortium U (2014) UniProt: a hub for protein information. Nucleic Acids Res 43: D204-D212.
- Choi Y, Chan AP (2015) PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31: 2745-2747.
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248.
- Karthik D, Majumder P, Palanisamy S, Khairunnisa K, Venugopal V, et al. ( 2014) Targeting cysteine rich C1 domain of Scaffold protein Kinase Suppressor of Ras (KSR) with anthocyanidins and flavonoids-a binding affinity characterization study. Bioinformation 10: 580-585.
- Capriotti E, Fariselli P, Casadio R (2005) I-Mutant 2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic acids Res 33: W306-W310
- Visualizer DS, v2. 5.1. 9167. Accelrys Software Inc.
- Li H, Leung KS, Nakane T, Wong MH (2014) iview: An interactive WebGL visualizer for protein-ligand complex. BMC Bioinformatics 15: 56.
- Li H, Leung KS, Ballester PJ (2014) Wong MH istar: A web platform for large-scale protein-ligand docking. PLoS One 9: e85678.
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, et al. (2015) Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 13: 1325-1335.
- Maunz A, Gütlein M, Rautenberg M, Vorgrimmler D, Gebele D, et al. (2013) Lazar: A modular predictive toxicology framework. Front Pharmacol 4: 38.
- Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R (2014) ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res 42: W53-W58.
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, et al. (2015) ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862-D868
- Vasan N, Boyer JL, Herbst RS (2014) A RAS renaissance: Emerging targeted therapies for KRAS-mutated non–small cell lung cancer. Clin Cancer Res 20: 3921-3930.
- Daina A, Michielin O, Zoete V (2017) SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7: 42717.
- Laskowski RA, Swindells MB (2011) LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51: 2778-2786.

