Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 10 Issue: 1
Fabrication of Paclitaxel and 17AAG-loaded Poly-É›- Caprolactone Nanoparticles for Breast Cancer Treatment
Yvonne Abbey Berko1, Funmilola A. Fisusi1,2 and Emmanuel O. Akala1*1 Center for Drug Research and Development, Department of Pharmaceutical Sciences, College of Pharmacy, Howard University, 2300 4th Street, NW, Washington, DC 20059, USA
2 Drug Research and Production Unit, Faculty of Pharmacy, Obafemi Awolowo University, Ile-Ife, Nigeria
*Corresponding Author : Emmanuel O. Akala
Center for Drug Research and Development,
Department of Pharmaceutical Sciences, College of Pharmacy, Howard University,
2300 4th Street, NW, Washington, DC 20059, USA
Tel: 2028065896
E-Mail: eakala@howard.edu
Received date: December 20, 2020; Accepted date: January 04, 2021; Published date: January 11, 2021
Citation: Berko YA, Fisusi FA, Akala EO (2021) Fabrication of Paclitaxel and 17AAG-loaded Poly-ɛ-Caprolactone Nanoparticles for Breast Cancer Treatment. J Pharm Drug Deliv Res 10:1.
Abstract
Objective: The aim of this study is to design, fabricate and determine the cytotoxic effects of dual loaded paclitaxel and 17-AAG in stealth polymeric nanoparticles. The nanoparticles were fabricated by dispersion polymerization. Methods: Two breast cancer cell lines (MCF-7 and SKBR-3) were cultured and treated with media only, blank nanoparticles, paclitaxel (as a free drug), 17-AAG (free drug), paclitaxel + 17- AAG combination (as free drugs), and paclitaxel + 17-AAG combination loaded in poly-�?-caprolactone stealth nanoparticles. Each drug in the combination was half the concentration of the single free drug. Results: The cytotoxic effects of the paclitaxel treatment and that of the combination (free drug) were found to be similar in both SKBR3 and MCF7 cell lines. Similar cytotoxic effects were observed for the drug combination both in the drug loaded nanoparticles formulation and in free drug form for both cell lines. Conclusion: Both paclitaxel and 17-AAG were effectively loaded and released from the polymeric nanoparticles. Paclitaxel (free drug), paclitaxel-17AAG combination (free drug), and dual drug-loaded nanoparticles had similar cytotoxic effects on both cell lines. Paclitaxel and 17-AAG combination resulted in synergistic effect: paclitaxel in the combination with 17-AAG was half its original concentration and yielded similar cytotoxic effect. The dose of paclitaxel was reduced without lowering its therapeutic efficacy
Keywords: Cytotoxicity; Breast cancer; SKBR-3; MCF-7; Nanoparticles; Paclitaxel; 17-AAG; Combination chemotherapy
Introduction
Taxanes (paclitaxel and docetaxel) have remarkable anticancer efficacy; they are among the bioactive agents established as first-line chemotherapeutics for the treatment of breast cancers. However, they have poor selectivity and high toxicity which are the most important factors for discontinuation of cancer chemotherapy [1]. Due to the poor solubility of paclitaxel and docetaxel, they are formulated as Taxol® and Taxotere® respectively for clinical use; the solvents and surfactants used in their preparations add to the toxic effects of taxanes during infusion [2]. In addition to toxicity, the clinical success of taxanes has been limited by the insurgence of cellular resistance, mainly mediated by expression of the multidrug resistant phenotype or by microtubule alterations [3, 4]. Paclitaxel acts at the G2/M phase of mitosis, causing mitotic arrest by binding to the beta-tubulin subunits of microtubules, stabilizing and protecting the microtubule polymer from disassembly [5]. This action disrupts the appropriate assembly of microtubules necessary for mitosis. The mechanism of action of paclitaxel also includes the production of macrophage IL-12 in tumor bearing hosts that downregulates the tumor growth significantly by selective dysregulation of IL-12 p40 expression; paclitaxel also reduces glycolysis by specific mechanisms [6,7].
Activation of the heat shock response is believed to be a general property of cancer cells involved in the initiation and maintenance of the transformed phenotype. A particularly important component in the heat shock response is heat shock protein 90 (HSP90). The multichaperone heat shock protein (HSP90) complex mediates the maturation and stability of a variety of proteins, many of which are crucial in oncogenesis including epidermal growth factor receptor (EGFR), HER2, AKT, Raf, p53, and cdk4 [8]. These proteins are referred to as “clients” of HSP90 and include proteins that contribute to all six “hallmarks of cancer” including self-sufficiency in growth signaling, insensitivity to antigrowth signals, evasion of apoptosis, sustained angiogenesis, tissue invasion and metastasis, and limitless replicative potential [9]. Inhibition of HSP90 function disrupts the complex and leads to degradation of client proteins in a proteasomedependent manner [10]. This disruption results in simultaneous interruption of many signal transduction pathways crucial to tumor progression and survival. Consistent with the role of an activated heat shock response in cancer, HSP90 is over-expressed in cancer cells; it is correlated with disease progression in melanoma and associated with decreased survival in breast cancer, gastrointestinal stromal tumors (GIST), and non-small cell lung cancer.
HSP90 inhibitors display remarkable selectivity for cancer cells as compared to normal cells and the inhibitors have been shown to accumulate in tumor tissue while being rapidly cleared from the circulation and normal tissues [11-14]. HSP90 inhibitors, such as 17- AAG and geldanamycin (GA), sensitize lung and breast cancer cells to paclitaxel induced cytotoxicity both in vitro and in vivo [15]. Further, concurrent exposure to 17-AAG and paclitaxel is required for the synergistic activity of the two drugs. 17-AAG has been shown to sensitize cancer cells to apoptosis stimulated by paclitaxel when both drugs are administered in combination. One of the mechanisms of paclitaxel resistance is by means of microtubule associated proteins such as Tau. Both paclitaxel and Tau bind to the beta-tubulin subunits of microtubules and stabilize them. The difference between the two is that when paclitaxel binds to the microtubule the affinity of the bond is so strong that there is no room for disassembly; whereas the binding affinity between Tau and microtubules permits the disassembly of the microtubule polymer resulting in mitosis. Due to the competition between Tau and paclitaxel for the same binding position on the microtubule, paclitaxel tends to be ineffective in the presence of aggregated Tau proteins [16, 17]. Tau protein is an HSP90 client protein and hyperphosphorylated Tau binds to HSP90 at a high affinity, resulting in aggregated Tau proteins. Microtubules at Tau aggregation sites are less susceptible to paclitaxel binding. Therefore, in the absence of heat shock protein 90, Tau protein aggregation is drastically reduced and paclitaxel-microtubule binding is increased, enhancing the therapeutic efficacy of paclitaxel in causing mitotic arrest. Since 17AAG is an HSP90 inhibitor, the combination of paclitaxel and 17AAG would greatly diminish paclitaxel resistance [18-21].
Although the combination of these two drugs seems promising in enhancing the efficacy of breast cancer treatment, there are challenges and limitations that pose as a threat to the efficiency of this combination when administered as free drugs in solution. These limitations include: the poor solubility of both drugs; lack of specificity (targetability); the use of cremophor EL to enhance solubility in both 17-AAG and paclitaxel which may induce hypersensitivity reactions [22]; incompatibility of cremophor EL to certain plastics which may be used as infusion bags; the physical instability of paclitaxel which causes the drug to precipitate during storage; and the complicated dosing regimen associated with this combination [23-25].
Therapeutic nanoparticles are capable of providing site-specific tumor targeting (active or passive targeting) thereby reducing toxicity in healthy cell, improving the solubility of anticancer drugs by incorporating them in the hydrophobic core [26, 27] and synchronizing the disposition (pharmacokinetics) of encapsulated (combination) drugs (varying biodistribution/pharmacokinetics of combination drugs through cocktail administration has been attributed to their ineffectiveness in the clinic) [28]. Further, therapeutic nanoparticles could overcome drug resistance due to P-glycoprotein efflux pump and enhance anticancer activity of therapeutic drugs (concurrent chemotherapy is more effective than sequential use of the chemotherapeutic agents). The stealth property of the nanoparticles due to PEG on the surface will prevent opsonization and capture by the reticuloendothelial system (RES). Dose reduction coupled with a better quality of life during chemotherapy would also improve patient compliance.
The use of biodegradable (poly-ɛ-caprolactone) nanoparticles in the delivery of 17-AAG – paclitaxel combination was investigated in this work as an excellent way to overcome the limitations associated with the administration of chemotherapeutic agents as free drugs in solution with the goal of enhancing breast cancer treatment.
Material and Methods
Materials
ɛ-caprolactone monomer (Sigma Aldrich, 99%) and toluene (Sigma, 99.9%) were distilled before use. Tin (II) 2-ethylhexanoate (stannous octoate), 2,4-dimethoxybenzaldehyde (DMBA) (98%), 2- Hydroxyethyl methacrylate (HEMA) (Sigma, 97%), paratoluenesulfonic acid monohydrate (Sigma>98.5%), anhydrous dichloromethane (DCM) (>99.8%), and triethylamine(TEA) (Sigma,>99%) were used as received. Poly (ethylene glycol) n monomethyl ether mono methacrylate (PEG-MMA, MW=1,000) was obtained from Polysciences, Inc. (Warrignton, PA, USA). Benzoyl peroxide (BPO), N-phenyldiethanolamine (N-PDEA) and acetone (HPLC grade) were obtained from Sigma Aldrich. Paclitaxel and 17AAG were obtained from LC Laboratories. SKBR3 cell line (American Type Culture Collection (ATCC) (Manassas VA, USA)), MCF7 cell line (Sigma Aldrich (St. Louis, MO, USA)), fetal bovine serum (FBS) (ATCC, Manassas VA, USA), penicillin/streptomycin (Fisher Scientific), Eagle’s minimum essential medium (EMEM) and McCoy’s 5A medium (ATCC, Manassas VA, USA), nonessential amino acid (NEAA) (Sigma Aldrich), trypsin EDTA (ATCC, Manassas VA, USA) were used in cell culture studies.
Methods
Synthesis and characterization of poly (Ɛ-caprolactone) macromonomer: HEMA and epsilon caprolactone monomer were dried over activated molecular sieves for 24h and distilled under negative pressure in an oil bath. Toluene was dried over calcium hydride for 1h prior to distillation. Silicone oil bath was heated and equilibrated to 120°C and 12.75 mL distilled Ɛ-caprolactone was polymerized in the presence of 2.8 mL distilled HEMA and 0.0375 mL of 0.4M stannous octoate by ring opening polymerization. Prior to polymerization, the mixture was placed under vacuum for 10 minutes without stirring. Subsequently it was lowered into the oil bath set at 120°C and stirred at 350 rpm. After 24h, the reaction was removed from the oil bath and allowed to cool for a few minutes, followed by the addition of 10 ml dichloromethane (DCM).
The product was added to 100 mL DCM and filtered through a Whatman filter paper using a vacuum pump and a Buchner funnel [29]. The filtrate was decanted into 1000 mL beaker after which cold hexane was added to precipitate the macromonomer. The final product was dried in the vacuum oven over phosphorous pentoxide. Characterization of poly (Ɛ-caprolactone) macromonomer was carried out as follows. Sample of the dried macromonomer was dissolved in chloroform-D for 1HNMR study to determine its purity and in tetrahydrofuran for gel permeation chromatography (GPC: Waters 2690 with a Waters 2410 differential refractive index detector) to determine the molecular weight. Polystyrene standards were used for calibration. The macromonomer was also characterized by Fourier transform infrared spectroscopy (FT-IR).
Synthesis and characterization of di (2-methacryloyloxyethoxy)- [2,4 dimethoxyphenyl] methane (DMDPM; pH sensitive crosslinker): HEMA was dried over activated molecular sieves for 24h and distilled under negative pressure in an oil bath before use. Molecular sieves were activated in the oven at 120°C for 3h and placed in a round bottom flask containing a magnetic stirrer together with a mixture of 3.9964g of 2,4-Dimethoxybenzaldehyde (DMBA), 12 mL distilled HEMA, and 0.725g para-toluene sulfonic acid monohydrate. Anhydrous dichloromethane ((DCM) 30 mL) was injected into the flask and stirred at room temperature under nitrogen gas for 30 minutes [30]. The reaction was left to run for 24 h; it was quenched in an ice bath by injecting 4.2 mL of triethylamine into the flask to neutralize the acidic catalyst used. It was stirred for 30 minutes at 0°C. The final product was filtered, washed with dichloromethane, evaporated, and purified by column chromatography. Aluminum oxide was used as the stationary phase; while the mobile phase consisted of hexane/ethyl acetate (6:1) with 1% (v/v) triethylamine. The crosslinker was analyzed by 1H-NMR, using a Brooker ADVANCE 400 MHz NMR spectrometer. Fourier-transform infrared analysis of the crosslinker was carried out using Perkin Elmer spectrum 100 FTIR spectrometer. Liquid chromatography mass spectrometry (LC-MS: Agilent model (1260) Infinity (Agilent Technologies, Palo Alto, USA)) was used to determine the molecular weight of the crosslinker.
Fabrication and characterization of dual-loaded nanoparticles: The dual-loaded nanoparticles were prepared using dispersion polymerization method as previously described [29-31]. Briefly, poly- ε-caprolactone macromonomer (0.28 mmol), PEG-MMA macromonomer (1.01 mmol) and the crosslinker (DMDPM; 0.373 mmol) were weighed separately and dissolved in acetone and transferred into a round bottom flask with a rubber closure. The solution was flushed with nitrogen gas with constant stirring at 200 rpm. N-phenyldiethanolamine (NPDEA) and benzoyl peroxide (BPO) [redox initiator system; (1:1, 0.594 mmol)] were injected into the flask after 10 and 20 minutes respectively of commencing the reaction. Paclitaxel and 17-AAG (0.0171 mmol each) were then injected into the reaction solution. The reaction was left to proceed at room temperature with constant stirring (200 rpm) and under the nitrogen atmosphere for 6h.The reaction was allowed to proceed for a total of 18h after initiation. The drug-loaded nanoparticles were purified by centrifugation using an ultracentrifuge. After centrifugation, the supernatant was removed and the nanoparticle was dispersed in deionized water and lyophilized for 24h and stored in the refrigerator at 4⁰C.
Particle size: The average particle size was determined by dynamic light scattering (DLS) technique using a Brookhaven 90 plus particle size analyzer. Samples of the nanoparticles were diluted with deionized water, probe sonicated for 45 seconds and filtered with a 5- micron syringe filter into a cuvette before the analysis. Polydispersity index of the particles (a measure of particles size distribution) was also determined.
Zeta potential: A known weight of freeze-dried nanoparticles was resuspended in deionized water, probe sonicated for 45 seconds and filtered through a 5-micron syringe filter. 1 mL of the filtrate was diluted with 1 mL deionized water and analyzed using a Brookhaven 90 plus, Zetaplus zeta potential analyzer. For each sample, 5 measurements were taken, and their average determined.
Zeta potential: A known weight of freeze-dried nanoparticles was resuspended in deionized water, probe sonicated for 45 seconds and filtered through a 5-micron syringe filter. 1 mL of the filtrate was diluted with 1 mL deionized water and analyzed using a Brookhaven 90 plus, Zetaplus zeta potential analyzer. For each sample, 5 measurements were taken, and their average determined.
Scanning electron microscopy (SEM) for nanoparticle morphology: Different dilutions of nanoparticle suspension in distilled water were placed on a carbon tape affixed to a specimen (aluminum) stub (SPI Supplies, Inc.) and dried in a vacuum oven for 24h. The samples were gold coated with Hummer sputtering machine for 2 minutes under argon atmosphere to improve conductivity. The samples were then observed using JEOL JSM 7600F scanning electron microscope. A 5kV accelerating voltage and secondary electron mode was used with a working distance of 12 mm for the morphological characterization [30].
Drug loading (DL): Analysis of drug loading, encapsulation efficiency and in vitro drug release was done by reversed phase high performance liquid chromatography (RP-HPLC) using the Agilent - Hewlett Packard 1100 Series High Performance Liquid Chromatography System equipped with Eclipse plus C18 (4.6 x 150 mm, 5 μm particle size) column kept at 35°C using 30:42:28 (methanol, acetonitrile, and water) as the mobile phase. A known weight (ANp) of freeze-dried nanoparticles was dissolved in acetonitrile, filtered with a 0.2-micron syringe filter and analyzed by HPLC. The developed calibration curve was used to determine the amount of drug in the solution (Asol). The mobile phase was the same as that used for the calibration curve [29-32]. Equation 1 below was used to determine the percent amount of drug loaded.
(%) DL=((Asol)/(ANp)) x 100% Eq. 1
Encapsulation efficiency (EE): Nanoparticle encapsulation efficiency was determined under the assumption that the total amount of drug encapsulated equals the initial amount of drug used for the nanoparticle synthesis (Aprep) minus the amount of drug found in the supernatant (Asup) after centrifugation. Percent (%) encapsulation efficiency was calculated with equation (2).
(%) EE=((Aprep)-(Asup))/(Aprep)) x 100% Eq. 2
Development of calibration curve: Both 17-AAG and paclitaxel were dissolved in ethanol at concentrations of 0.5 μg/mL, 2.5 μg/mL, 5 μg/mL, 10 μg/mL, 20 μg/mL, 50 μg/mL and 100 μg/mL and analyzed by reversed phase high performance liquid chromatography (RP-HPLC) (Agilent-Hewlett Packard 1100 series) equipped with Eclipse plus C18 (4.6 x 150 mm, 5 μm particle size) column kept at 35°C. Methanol, acetonitrile, and water (all HPLC grade) were filtered using 0.2-micron nylon filter paper and used at a 30:42:28 ratio respectively as the mobile phase. 20μL was injected and analyzed at a flow rate of 1 mL/min (λ max for paclitaxel is 227 nm and λ max for 17AAG is 334 nm [33,34].
Drug release studies: In vitro drug availability of paclitaxel and 17AAG was studied using a dialysis method. A known weight of freeze-dried nanoparticles was suspended in 2 mL acetate buffer (pH 5.0) and placed in a dialysis bag (12-14k Da MWCO (Spectra/ Por®CE)). The dialysis bag was then placed in a 15 mL Eppendorf tube containing the release medium consisting of acetate buffer (pH 5.0) and 0.1% w/v polysorbate 80 (Tween 80) [35]. The tube was clamped to a Labquake® shaker capable of 360⁰ rotation at 37oC in an endotherm laboratory oven (Fisher Scientific, USA). At specific time intervals, an aliquot of the release medium was taken and replaced with same amount of fresh medium each time to maintain constant volume [29-32]. Studies were done in triplicates. All samples taken were filtered with a 0.2 micron syringe filter and then analyzed by HPLC to determine the concentration of each drug.
Cell Cultures
SKBR3 cells were cultured in McCoy’s 5A medium supplemented with fetal bovine serum (FBS; 10%v/v), and penicillin/streptomycin [penicillin G (100U/mL) and streptomycin sulfate (100μg/mL); 1% v/v]. MCF7 cells were cultured in Eagle’s minimum essential medium (EMEM) supplemented with L-glutamine (2mm) nonessential amino acid [(NEAA), 1% v/v], fetal bovine serum [(FBS), 10%v/v] and penicillin/streptomycin [penicillin G (100U/mL) and streptomycin sulfate (100μg/mL); 1% v/v]. Cells were maintained in a humid incubator (5% CO2, at 37oC). The cells were maintained in culture and split when 75-80% confluence was reached in the culture flask.
Cytotoxicity Experiments
Cells were seeded in 96-well microplates (5,000 cells in 100 μl of culture medium) and incubated for 24 h to allow the cells to attach. The culture medium was then removed from the wells and replaced with fresh medium containing different concentrations of the drugs (paclitaxel, 17-AAG or equimolar combination of both drugs with the concentration of paclitaxel and 17-AAG halved) in solution or drug loaded nanoparticles. The treatments were applied at concentrations ranging from 0.1nM to 1000nM. Some cells (controls) were treated with culture medium only, culture medium with 0.025% DMSO and culture medium containing blank polymer nanoparticles. For treatment with the drugs in solution, the drugs were weighed separately into glass scintillation vials and dissolved in DMSO and culture medium to produce an initial stock of 10μM which was diluted to 1000nM. The 1000nM solution was then used to prepare the dilutions used for treating the cells. Equal volumes of the single drug dilutions were mixed (1:1) to prepare drug solutions for the combination drugs treatment (the concentration of each single drug solution was halved in the combination drug solution). For dual loaded nanoparticles (DLNP), the drug loading of paclitaxel in the nanoparticles (1.71%) was used to determine the quantity of nanoparticles to use for cell treatment.
To prepare the initial stock solution for treatment, DLNP was weighed into a glass scintillation vial and suspended in culture media to obtain an initial concentration of 10μM. This was further diluted down to obtain the highest treatment concentration (1000nM), from which serial dilutions were prepared to obtain the solutions for treating the cells. The blank polymer nanoparticle formulation was also weighed and similarly diluted to obtain the concentration of nanoparticles corresponding to what is contained in the highest concentration (1000nM) of DLNP. Subsequently, cell plates were assayed at predetermined time points of 24 h, 48 h, 72 h, 96 h and 120 h after exposure of the cells to the treatment solutions to determine cell viability.
At each time point, the treatment media were removed from the cell plates and fresh media were added. An equal volume of CellTiter- Glo® luminescence assay reagent (which produces luminescence signal proportional to the amount of ATP generated) was then added to each well and the plate was shaken for 2 minutes [(Clariostar Plus Microplate Reader, BMG Tech) equipped with an in-built shaker] to lyse the cells. The plates were kept in the dark for 30 minutes, after which luminescence was measured as a surrogate for the number of viable (metabolically active) cells present.
Results and Discussion
The macromonomer was synthesized by ring opening polymerization of the epsilon caprolactone monomer in the presence of HEMA as the initiator [Scheme 1]. 1HNMR spectrum displays (spectrum not shown) olefinic protons of a C=C bond at chemical shift 5.6 ppm and 6.1 ppm, showing the incorporation of HEMA. The gel permeation chromatography (GPC) data showed a single peak (indicating its purity), with a polydispersity index (PDI) of 1.63. Number average molecular weight (Mn) calculated by 1HNMR method was 1075g/mol; while that of GPC was 1269g/mol. The weight average molecular weight determined by GPC was 2074g/mol.

Scheme 1: Ring-opening polymerization scheme for the synthesis of poly (ε -caprolactone) macromonomer.
It is usual to have different Mn values of the same polymer determined by 1H NMR analysis and by GPC [29].It has been ascribed to the use of polystyrene standards for calibration. It is due to differences in the hydrodynamic volume of polystyrene relative to poly-caprolactone-HEMA macromonomer. FT-IR spectrum (data not shown) reveals the presence of significant functional groups such as C-H: 2943.79 cm-1, C=O: 1721.91 cm-1 and the C=C at 1636.91 cm-1.
The early endosome has a pH of about 5-6; while the lysosome has a pH of about 4-5 [30-36]. Consequently, the purpose of incorporating an acid-labile crosslinker into the design of our nanoparticles was to facilitate the design of nanoparticles that will be stable in blood circulation but degrade in the acidic environment. Our laboratory [30] has reported the synthesis of three different types of acetal crosslinkers using three different benzaldehydes which differ in the number of methoxy groups present on the aromatic ring.
In this work, we synthesized the pH-sensitive acetal crosslinker with two methoxy groups present on the aromatic ring and degradable in an acidic environment. The crosslinker was characterized by 1H NMR, FT-IR and liquid chromatography mass spectrometry (LC-MS) and used in this work. The 1H NMR spectrum (data not shown) displays the acetal proton peak where the crosslinker hydrolyzes in an acidic medium as a singlet at chemical shift 5.84pp, and methoxy groups at 3.80ppm and 3.81pp, as expected [30]. Also, the FT-IR spectrum shows the presence of important functional groups as follows: The C-H group at 2955.08 cm-1, Carbonyl group (C=O) at 1715.39 cm-1, C=C group at 1636.92 cm-1 and aromatic ring between 1376.62 cm-1 – 1613.76 cm-1 [30]. According to the chemical formula of the crosslinker (C21H28O8), the expected molecular weight was 408.4422 g/mol. The molecular weight of the synthesized crosslinker determined by liquid chromatography mass spectroscopy was 408.1775g/mol [Figure 1].

Figure 1: The molecular weight of the synthesized crosslinker, LCMS of di (2-methacryloyloxyethoxy)-[2,4-dimethoxyphenyl]methane (DMDPM; pH Sensitive Crosslinker) Expected MW(C21H28O8)=408.4422g/mol. Molecular weight observed in spectrum=MW(C21H28O8)+MW(Na+)=431.1673g/mol Obtained=431.1673g/mol –22.9898g mol =408.1775g/mol.
We implemented a central composite face-centered statistical experimental design (CCF) in three independent factors and seventeen runs for the fabrication nanoparticles by dispersion polymerization technique, followed by optimization as shown in Table 1[37].
Table 1: Optimization of dual-loaded (Paclitaxel and 17-AAG) nanoparticles.
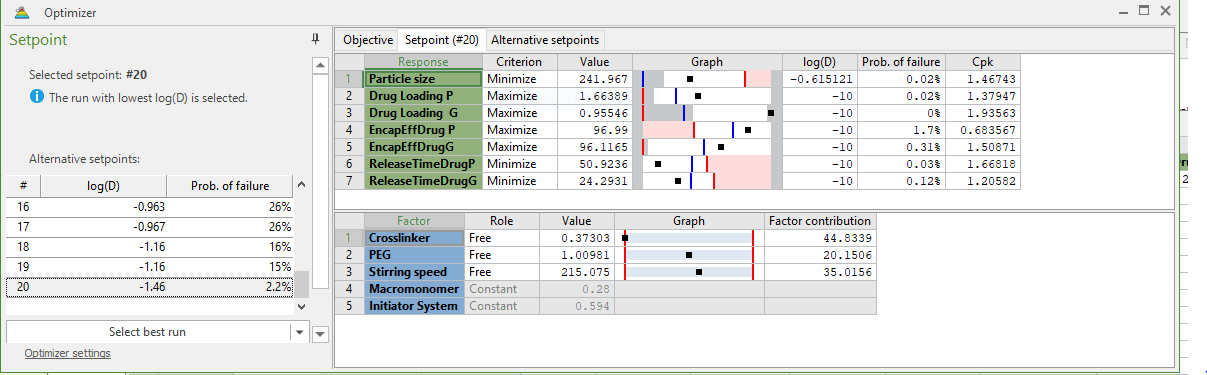
The computer software optimizer was used for optimization to select the factor combination to minimize the particle size, to minimize the time (h) for maximum release of paclitaxel and 17-AAG, to maximize paclitaxel and 17-AAG loading efficiency and to maximize paclitaxel and 17-AAG encapsulation efficiency. The combination of factors to give optimal formulation is shown in Table 1 (Crosslinker 0.373 mmol, PEG 1.00981 mmol, stirring speed, 215.075 rpm (approximated to 200 rpm in this work), macromonomer 0.28 mmol and initiator system, 0.594 mmol) (see fabrication of nanoparticles). Figure 2 shows the average particle size and particle size distribution (243.6 ± 0.50 nm) with a polydispersity index of 0.192 ± 0.084). Figure 3 shows the scanning electron micrographs (morphology of the nanoparticles).

Figure 2: Particle size analysis data of dual-loaded (Paclitaxel and 17-AAG) nanoparticles by dynamic light scattering. The average nanoparticle diameter recorded is 243.6 ± 0.50 nm with a polydispersity index of 0.192 ± 0.084.

Figure 3: Scanning electron micrographs of dual-loaded nanoparticles.
Physicochemical characteristics of nanoparticles such as particle size and surface charge play an important role in determining their in vitro drug release as well as their in vivo pharmacokinetics and biodistribution, and hence the therapeutic efficacy of the encapsulated drug [38,39]. When large colloidal particles are administered intravenously they are rapidly taken up by reticuloendothelial system (RES) while small particles and those with a hydrophilic surface show slow clearance rate [40]. In this study, the hydrophilic surface was provided by PEG-MMA, which has been shown to promote long circulation of nanoparticles. Figure 4 shows the negative zeta potential of the nanoparticles (-35 ± 3.69 mv). Zeta potential is a measure of the effective electric charge on the surface of the nanoparticles. Thus, negatively charged nanoparticles will repel each other, making them less likely to aggregate in suspension [41]. The tendency of cells to internalize nanoparticles greatly depends on the overall surface charge of surrounding fluids which arises from the cell-nanoparticle surface charge interactions and studies have shown that nanoparticle surface charge influences cellular uptake. Higher cellular uptake and lower protein adsorption was detected with negatively charged more than with positively charged nanoparticles [42].

Figure 4: Zeta potential of dual-loaded (Paclitaxel and 17-AAG) nanoparticles.
The average zeta potential is -35 ± 3.69 mV.
Figure 5 shows in vitro availability of the dual-loaded nanoparticles. The average in-vitro release studies revealed a maximum release time of 53h for paclitaxel and 30h for 17-AAG.Data obtained from HPLC analysis showed that drug loading and encapsulation efficiency for paclitaxel were 1.71% ± 0.03 and 93.3% ± 0.03 respectively; while for 17-AAG, drug loading and encapsulation efficiency were 0.92% ± 0.06 and 97.83% ± 0.01 respectively.

Figure 5: In vitro drug release isotherm of dual-loadednanoparticles (Paclitaxel: ɛ=0.06-0.314; n=3; 17-AAG: ɛ=0.08-0.646; n=3).
The result of cytotoxicity experiments showed that there was no appreciable cytotoxic effect in both cell lines at the 24 h time point with cell viability of approximately 100% across all the treatments at all tested concentrations (Figure 6).

Figure 6: Comparison of the effects of all the treatment arms on SKBR3 and MCF7 cell lines after 24 hours of cell exposure to treatment (DLNP=Combination drug loaded nanoparticles, PTX=Paclitaxel solution, 17AAG=17AAG solution and PTX +17AAG= combination drug solution at half concentration of each drug).
Although there was a noticeable decrease in the viability of SKBR3 cells treated with paclitaxel and the combination with 17-AAG (drugs in solution) at the 48 h time point, the viability was still above 60% in all tested concentrations (Figure 7). The drug loaded nanoparticle formulation also produced similar effects at higher (10nM-1000nM) concentrations. The MCF7 cells, on the other hand, did not show appreciable response at this time point, with viability of above 75% at all tested concentrations of the treatments applied. At 48 h, it is becoming discernible that SKBR3 cells are more responsive to the treatments than MCF7 cells.

Figure 7: Comparison of the effects of all the treatment arms on SKBR3 and MCF7 cell lines after 48 hours of cell exposure to treatment (DLNP=Combination drug loaded nanoparticles, PTX=Paclitaxel solution, 17AAG=17AAG solution and PTX+ 17AAG=combination drug solution at half concentration of each drug).
17AAG=combination drug solution at half concentration of each drug).
In SKBR3 cells at 72 h time point, there was appreciable reduction in cell viability across all tested concentrations, with viability lower than 50% for all applied treatments except 17-AAG (Figure 8).

Figure 8: Comparison of the effect of all the treatment arms on SKBR3 and MCF7 cell lines after 72 h of cell exposure to treatment (DLNP=Combination drug loaded nanoparticles, PTX=Paclitaxel solution, 17AAG=17AAG solution and PTX+17AAG=combination drug solution at half concentration of each drug).
Cells treated with 17-AAG showed reduction in viability only at concentrations of 100nM and 1000nM. For MCF7 cells (Figure 8), viability was still in the order of 60% or higher across all treatments applied. A similar pattern was found at the 96h time point with cell viability below 50% in SKBR3 cells (down to about 20%) in all applied treatments apart from 17-AAG, which resulted in viability of about 50% at 100nM and 1000nM. In MCF7 cells viabilities were still up to 50% for all applied treatments (Figure 9).

Figure 9: Comparison of the effects of all the treatment arms on SKBR3 and MCF7 cell lines after 96 hours of cell exposure to treatment ((DLNP = Combination drug loaded nanoparticles, PTX=Paclitaxel solution, 17AAG=17AAG solution and PTX +17AAG=combination drug solution at half concentration of each drug).
At the 120 h time point, all treatments (including 17-AAG at 100nM and 1000nM) produced viability lowers than 50% in SKBR3 cells (Figure 10). In MCF7 cells, at 1nM concentration, paclitaxel was superior to all other treatments; while drug combinations (paclitaxel and 17-AAG) in nanoparticles and in solution have the same behavior (similar level of cytotoxicity). 17-AAG does not show much cytotoxicity. Paclitaxel, DLNP and paclitaxel combination with 17- AAG (drugs in solution) produced viability lower than 50% at concentrations from 10nM to 1000nM; while 17-AAG (drug in solution) produced viability lower than 50% only at the highest tested concentration of 1000nM (Figure 10).

Figure 10: Comparison of the effects of all the treatment arms on SKBR3 and MCF7 cell lines after 120 hours of cell exposure to treatment (DLNP=Combination drug loaded nanoparticles, PTX=Paclitaxel solution, 17AAG=17AAG solution and PTX + 17AAG=combination drug solution at half concentration of each drug).
From the data, it can be deduced that 17-AAG does not seem to exert considerable cytotoxic effect on its own. However, 17-AAG appears to be contributing to the cytotoxic effect of paclitaxel, as the combination treatment (PTX+17-AAG) containing half the concentration of each drug (the drug solutions for combination drug treatment were prepared by mixing the solutions of the individual drugs in a 1:1 ratio for each concentration with the concentration halved) produced similar cytotoxic effect as the whole concentration of paclitaxel. The same trend is seen in DLNP. Thus, although the concentration of paclitaxel and 17-AAG had been reduced by half in the combination, similar levels of cytotoxicity were produced in treated cells. The data support previous results indicating that 17-AAG sensitized the cytotoxicity of paclitaxel [18-21]. The therapeutic potential of HSP90 inhibition is being evaluated extensively in a number of clinical trials, including 17-AAG which is now in phase III clinical trials. Additionally, inhibition of HSP90 function by 17-AAG has been reported to enhance the apoptotic effects of cytotoxic agents such as paclitaxel and doxorubicin. A recent review highlights the extensive clinical evaluations that 17-AAG has undergone as both a single agent and in combination with approved therapies such as bortezomib, imatinib, docetaxel, paclitaxel, irinotecan, sorafenib, trastuzumab, cisplatin, cytarabine, rituximab, etc. with enhancement or synergistic effects [43].
In order to understand the concurrent effects of the drugs in solution for the current combination (paclitaxel: 17-AAG; 1:1), the effect produced was assessed by computing the combination index (CI) by the Chou-Talalay method, using the Compusyn® software. In the Chou-Talalay method, CI values less than 1 indicate synergism, CI values equal to 1 indicate summation, while CI values greater than 1 indicate antagonism [44, 45]. The CI for MCF7 cells at the ED50 were 0.27, 0.34 and 0.58 at the 48, 72 and 96h time points respectively over the concentration range from 0.1 – 1000nM. These values are less than 1 suggesting a synergistic effect for the cytotoxic activities of the current drug combination in MCF7 cells. The CI values for SKBR3 could not be computed because the viability was well below 50% at the time points greater than 48 h and there was no distinct difference in viability across all tested concentrations at those time points.
Statistical analysis: To show the synergistic effects of the dualloaded nanoparticles and the drug solutions on SKBR3 cell lines, statistical analysis was carried out on the data obtained at 96 hours. Two factor (drug concentration and type of treatment) analysis of variance (ANOVA) with F-test of significance was carried out on the data of % viability of SKBR3 HER2-positive cancer cell lines at 96h using Design-Expert® software (Version 12.0). Pair wise comparisons test (Tukey’s test) was used to locate specific differences between the means. The level of significance was set at 0.05.
The analysis of variance (ANOVA) test was conducted to determine whether the difference in % viability among the types of treatment was statistically significant, and also to determine the effect of drug concentration on % viability.
The analysis of variance test (Table 2) shows that the model is significant (p < 0.0001), meaning that either one of the two factors (type of treatment or drug concentration) or both significantly affect % viability data. Further, the effect of type of treatment is significant (p<0.0001) showing that there is a statistically significant difference in the % viability among the four types of treatment (Paclitaxel solution (PTX), 17-AAG solution (17-AAG), Paclitaxel+17-AAG solution each at half the concentration of individual drug solution and Dual loaded nanoparticles (DLNP) containing the same drug concentration as PTX+ 17-AAG). The effect of drug concentration is also significant (p< 0.0001), showing that % viability of SKBR3 HER2-positive cancer cell line differs at different drug concentrations. The interaction of type of treatment with drug concentration is also significant, indicating that % viability of the type of treatment is not the same at different levels of drug (drug concentration). Figures 9 and 11 support the inferences.
Table 2: Analysis of variance (ANOVA) table for the effect of treatment [(Paclitaxel solution (PTX), 17-AAG solution (17-AAG), Paclitaxel+17-AAG solution each at half the concentration of individual drug solution and dual loaded nanoparticles (DLNP) containing the same drug concentration as PTX + 17-AAG)] and drug concentration on % viabilitySKBR3 cell line at 96 hours.
| Source | Sum of Squares | df | Mean Square | F-value | p-value | |
|---|---|---|---|---|---|---|
| Model | 1.20E+05 | 23 | 5200.54 | 377.73 | < 0.0001 | significant |
| A-Treatment | 29085.81 | 3 | 9695.27 | 704.19 | < 0.0001 | significant |
| B-Concentration | 61939.1 | 5 | 12387.8 | 899.76 | < 0.0001 | significant |
| Interaction (AB) | 28587.6 | 15 | 1905.84 | 138.43 | < 0.0001 | significant |
| Pure Error | 1321.72 | 96 | 13.77 | |||
| Cor Total | 1.21E+05 | 119 |

Figure 11: 3D plot showing the effect of type of treatment (PTX=Paclitaxel solution, 17AAG =17AAG solution and PTX +17AAG=combination drug solution at half concentration of each drug and DLNP=combination drug loaded nanoparticles containing half the concentration of each drug).
Having shown that ANOVA test is valid for our data on % viability of the treatments on SKBR3 HER2-positive cancer cell lines (validation data not shown), it is of interest to use multiple comparisons test (Tukey’s test) to compare the means of % viability for the treatments at each concentration. When the interaction of factors is significant, as we have in our ANOVA analysis (Table 2), comparisons between the means of a factor (i.e. type of treatment) may be obscured by the interaction of type of treatment with drug concentration. One approach to this situation is to fix one factor (i.e., drug concentration) at a specific level and apply Tukey’s test to the means of each type of treatment [30,46]. Tukey’s test makes use of studentized range and is applicable to pair wise comparison of means.
It requires a single range value for judging the significance of all differences.
The procedure involves computing a critical value, as shown in Equation 3, and applying it to differences between all pairs of means.
Tα = qα (p, fe) (MSE/n)0.5 Equation (3)
Where Tα is the Tukey’s critical value, qα is obtained from percentage points of the studentized range statistic; p is equal to t (the number of treatments), and fe is error degree of freedom.
From Equation (3), the Tukey’s critical value for the types of treatment was calculated as shown below.
T0.05 = q0.05 (4, 96)(13.77/4)0.5= 6.86
The level of significance was set at α= 0.05. The results are shown in Table 3.
Table 3: Tukey's test for the % viability studies of the four types of treatment at different drug concentrations.
| Difference in the means of % cell viability | Comparison with T0.05 | Conclusion |
|---|---|---|
| Tukey's test for the % viability studies of the four types of treatment at 0 nM (medium or blank nanoparticles). | ||
| Mediu-DLNP=2.31 | 2.31<6.86 | Not significant |
| Tukey's test for the % viability studies of the four types of treatment at 0.1nM. | ||
| PTX-(17-AAG+ PTX)=3.56 | 3.56<6.86 | Not significant |
| PTX-DLNP=75.08* | 75.08>6.86 | Significant |
| 17-AAG-PTX=60.63* | 60.63>6.86 | Significant |
| (17-AAG+PTX) - DLNP = 66.69* | 66.69>6.86 | Significant |
| 17-AAG-(17-AAG+PTX)=57.07* | 57.07>6.86 | Significant |
| 17-AAG -DLNP=14.61* | 14.61>6.86 | Significant |
| Tukey's test for the % viability studies of the four types of treatment at 1nM. | ||
| PTX-(17-AAG+PTX)=1.46 | 1.46<6.86 | Not significant |
| PTX-DLNP=71.84* | 71.84>6.86 | Significant |
| 17-AAG-PTX =54.31* | 54.31>6.86 | Significant |
| (17-AAG+PTX) - DLNP=70.42* | 70.42>6.86 | Significant |
| 17-AAG-(17-AAG+PTX)=52.85* | 52.85>6.86 | Significant |
| 17-AAG-DLNP=17.53* | 17.53>6.86 | Significant |
| Tukey's test for the % viability studies of the four types of treatment at 10 nM. | ||
| PTX-(17-AAG+PTX)=6.02 | 6.02<6.86 | Not significant |
| PTX - DLNP = 5.92 | 5.92<6.86 | Not significant |
| 17-AAG-PTX=54.56* | 54.56>6.86 | Significant |
| (17-AAG+PTX)-DLNP=0.11 | 0.11<6.86 | Not significant |
| 17-AAG - (17-AAG +PTX=48.54* | 48.54>6.86 | Significant |
| 17-AAG - DLNP = 48.65* | 48.65>6.86 | Significant |
| Tukey's test for the % viability studies of the four types of treatment at 100 nM. | ||
| PTX -(17-AAG+ PTX)=5.35 | 5.35<6.86 | Not significant |
| PTX-DLNP=2.24 | 2.24<6.86 | Not significant |
| 17-AAG-PTX=27.12* | 27.12>6.86 | Significant |
| (17-AAG+PTX-DLNP=3.12 | 3.12<6.86 | Not significant |
| 17-AAG-(17-AAG+PTX)=21.77* | 21.77>6.86 | Significant |
| 17-AAG-DLNP=24.89* | 24.89 > 6.86 | Significant |
| Tukey's test for the % viability studies of the four types of treatment at 1000 nM. | ||
| PTX-(17-AAG+PTX)=0.03 | 0.03<6.86 | Not significant |
| PTX-DLNP=0.81 | 0.81<6.86 | Not significant |
| 17-AAG-PTX=27.51* | 27.51>6.86 | Significant |
| (17-AAG+PTX)- DLNP=0.84 | 0.84<6.86 | Not significant |
| 17-AAG - (17-AAG +PTX)=27.23* | 27.23>6.86 | Significant |
| 17-AAG-DLNP =28.32* | 28.32>6.86 | Significant |
Table 3 shows that there is nostatistical significant different between the means of % viability of the blank nanoparticles and the medium (0 nM) indicating that the blank nanoparticles were not toxic. From 0.1nM to 1000nM concentrations, paclitaxel solution (PTX) and PTX+17-AAG (Paclitaxel+17-AAG solution, each drug at half the concentration of paclitaxel solution) showed the same effect on % viability of SKBR3 cell line. The implication is that 17-AAG is synergistic with paclitaxel. This synergistic effect becomes discernible for DLNP (dual loaded nanoparticles containing the same drug concentration as PTX+17-AAG) from 10 nM to 1000nM (Table 3 and Figure 11).
Combination therapies are beneficial and they potentially offer several advantages which include improvements in the toxicity profile (since lower concentrations of individual agents can be used), reduced or delayed development of drug resistance and improved efficacy [45,47]. Due to these advantages, combination therapies have become a standard for the treatment of several diseases and continue to represent a promising approach in indications of unmet medical need [47]. Drug-loaded nanoparticles provide an excellent platform for the application of combination therapy as they offer some benefits over conventional drug delivery systems. These advantages include the ability to carry multiple agents within the same nanoparticle system.
Thus for combination therapy, the agents to be delivered can be encapsulated within the same delivery system. Other advantages include the potential for controlled and site-specific delivery, lower dosage regimen, reduction in administered dose and toxic effects [48-51].This study has demonstrated that the application of paclitaxel and 17-AAG in the current combination would potentially afford a reduction in the dose (by half) required to produce the desired therapeutic effect and would thus lead to a reduction in toxicity. The use of the nanoparticle system would further extend the advantages by providing the possibilities for controlled and/or site-specific delivery to affected tissue or organ systems using the PEG molecules on the surface of the nanoparticles for tethering or conjugating ligands on the surface of the nanoparticles which can bind to the receptors expressed on the surface of cancer cells.
SKBR3 cells were found to be more sensitive to paclitaxel-17-AAG combination than MCF7 cells. It is known that SKBR3 cell line is HER-2-overexpressing. Data from 17-AAG clinical studies in which trastuzumab was combined with 17-AAG showed activity in patients with HER2-positive metastatic breast cancer who had progression of disease on trastuzumab therapy [10,52-54]. Moreover, HSP90 inhibitors such as 17-AAG potently downregulate the cell surface HER2. Combined 17-AAG and trastuzumab treatment induced synergistic growth arrest and cell death specifically in HER2- overexpressing but not in HER2-low breast cancer cells [54].
Figures 12 and 13 shows the effects of DLNP with time on both cell lines. It was observed that at 0.1nM and 1nM, there was no significant decrease in % cell viability within all the treatment times for both cell lines. This result may be attributed to the fact that low amounts of the drugs were loaded in nanoparticles. Cytotoxic effect occurred between 1nM and 10nM DLNP to varying extents in both cell lines and across all treatment times, after which the effect plateaued at higher concentrations. This plateau trend is typical of taxanes as reported in literature.

Figure 12: Effect of combination drug loaded nanoparticles treatment duration on the % cell viability of SKBR3 cells (n=5).

Figure 13: Effect of treatment duration of dual-loaded nanoparticles on the % cell viability of MCF7 cells (n=5).
Conclusion
The dispersion polymerization method was successfully used to fabricate an optimized dual drug-loaded nanoparticle formulation using a pH-sensitive crosslinker and a macromonomer. In-vitro cytotoxicity studies revealed that the blank nanoparticles were biocompatible with and non-toxic to the cells as compared to the control (media only). Also, cytotoxic effects of the paclitaxel treatment and that of the drug combination (free drugs or nanoparticles (DLNP) observed were similar in both SKBR3 and MCF7 cell lines suggesting synergistic or potentiation effects. Also, since paclitaxel in the combination is half its original concentration, and still yielded similar cytotoxic effect, we have been able to reduce the dose of paclitaxel without lowering its therapeutic efficacy. 17-AAG on its own was not as effective as compared to paclitaxel alone or in combination with paclitaxel.
Acknowledgement
Research reported in this publication was supported by NCI/NIH Grant #: 1SC1CA199810-01 awarded to Emmanuel O. Akala. This work was carried out in facilities supported by NCRR/NIH Grants #1C06 RR 020608-01 and #1 C06 RR 14469-01.
References
- Gastaldi D, Zonari D, Dosio F (2011) Targeted Taxane Delivery Systems: Recent Advances. Drug Delivery Letters 1: 105-117.
- Rowinsky EK, Donehower RC, Wood AJJ (1995) Paclitaxel (Taxol). N Eng J Med 335: 1004-1014.
- Greenberger LM, Williams SS, Horwitz SB (1987) Biosynthesis of heterogeneous forms of multidrug resistance-associated glycoproteins. J Biol Chem 262: 13685-13689.
- Gallett E, Magnani M, Renzulli ML, Botta M (2007) Paclitaxel and docetaxel resistance: Molecular mechanisms and development of new generation taxanes. Chem Med Chem 2: 920-942.
- Maresca TJ, Salmon ED (2010) Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. Journal of cell science 123: 825-835.
- Mullins DW, Burger CJ, Elgert KD (1999) Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. Journal of immunology 162: 6811-6818.
- Glass-Marmor L, Beitner R (1999) Taxol (paclitaxel) induces a detachment of phosphofructokinase from cytoskeleton of melanoma cells and decreases the levels of glucose 1,6-bisphosphate, fructose 1,6-bisphosphate and ATP. European journal of pharmacology 370: 195-199.
- Zhang H, Burrows F (2004) Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med 82:488-499.
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57-70.
- Lu X, Xiao L, Wanga L, Ruden DM (2012) Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochemical Pharmacology 83: 995-1004.
- McCarthy MM, Pick E, Kluger Y, Gould-Rothberg B, Lazova R, et.al. (2008) HSP90 as a marker of progression in melanoma. Ann Oncol 19: 590-594.
- Pick E, Kluger Y, GiltnaneJM, Moeder C, Camp RL, et, al. (2007) High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res 67: 2932-2937.
- Gallegos Ruiz MI, FloorK, Roepman P, Rodriguez JA, Meijer GA et, al. (2008) Integration of gene dosage and gene expression in non-small cell lung cancer, identification of HSP90 as potential target. PLoSOne 5.
- Li CF, Huang WW, Wu JM, Yu SC, Hu TH, et al., (2008) Heat shock protein 90 overexpression independently predicts inferior disease-free survival with differential expression of the alpha and beta isoforms in gastrointestinal stromal tumors. Clin Cancer Res 14: 7822-7831.
- Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N (2003) Inhibiton of heat shock protein 90 function down-regulates Akt kinase and sentizes tumors to Taxol. Cancer Res 63: 2139-2144.
- Kar S, Fan J, Smith MJ, Goedert M, Amos LA (2003) Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. The EMBO Journal 22: 70-77.
- Smoter M, Bodnar L, Duchnowska R, Stec R, Grala B, et al., (2011) The role of Tau protein in resistance to paclitaxel. Cancer chemotherapy and pharmacology 68: 553-557.
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, et al., (2007) The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J clin Invest 117: 648-658.
- Dickey CA, Dunmore J, Lu B, Wang JW, Lee WC et al,. (2006) HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB Journal : official publication of the Federation of American Societies for Experimental Biology 20: 753-755.
- Luo W, Dou F, Rodina A, Chip S, Kim J, et al., (2007) Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci USA 104: 9511-9516.
- Salminen A, Ojala J, Kaarniranta K, Hiltunen M, Soininen H (2011) Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer's disease. Prog neurobiol 93: 99-110.
- Mayer LD, Janoff AS (2007) Optimizing combination chemotherapy by controlling drug ratios. Mol Interv 7: 216-223.
- Mazzo DJ, Nguyen-Huu JJ, Pagniez S, Denis P (1997) Compatibility of docetaxel and paclitaxel in intravenous solutions with polyvinyl chloride infusion materials. American Journal of Health-System Pharmacy : AJHP : Official Journal of the American Society of Health-System Pharmacists 54: 566-569.
- Donyai P, Sewell GJ (2006) Physical and chemical stability of paclitaxel infusions in different container types. Journal of Oncology Pharmacy Practice : official publication of the International Society of Oncology Pharmacy Practitioners 12: 211-222.
- Finley RS, Balmer CM (1998) Concepts in Oncology Therapeutics (Mar 15, 1998)Published by American Society of Health-System Pharmacists Bethesda MD.
- Sparreboom A, Baker SD, Verweij J (2005) Paclitaxel Repackaged in an Albumin-Stabilized Nanoparticle: Handy or Just a Dandy? J Clin Oncol 23: 7765-7767.
- Emmanuel OA, Adesina SK (2017) “Fabrication of polymeric core-shell nanostructures” in Nanoscale Fabrication, Optimization, Scale-up and Biological Aspects of Pharmaceutical Nanotechnology.
- Hu C-MJ, Zhang L (2012) Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochemical Pharmacology 83: 1104-1111.
- Ogunwuyi O, Adesina S, Akala EO (2015) D-Optimal mixture experimental design for stealth biodegradable crosslinked docetaxel-loaded poly-epsilon-caprolactone nanoparticles manufactured by dispersion polymerization. Die Pharmazie 70: 165-176.
- Puri R, Berhe S, Akala E (2017) pH-sensitive polymeric nanoparticles fabricated by dispersion polymerization for the delivery of bioactive agents. Pharm Nanotechnol 3: 44-66.
- Adesina S, Wight S, Akala E (2014) Optimization of the fabrication of novel stealth PLA based nanoparticles by dispersion polymerization using D-optimal mixture design. Drug Dev Ind Pharm 40: 1547-1556.
- Akala EO, Adesina S, Ogunwuyi O (2016) Computer Optimization of Biodegradable Nanoparticles Fabricated by Dispersion Polymerization. Int J Environ Res Public Health 13: 47-63.
- Xia XJ, Peng J, Zhang PX, Jin DJ, Liu YL (2013) Validated HPLC Method for the Determination of Paclitaxel-related Substances in an Intravenous Emulsion Loaded with a Paclitaxel-Cholesterol Complex. Indian J pharm sci 75: 672-679.
- Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, et al., (2005) Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer research 65.
- Pradhan R, Poudel BK, Choi JY, Choi IS, Shin BS (2015) Erratum to: Preparation and evaluation of 17-allyamino-17-demethoxygeldanamycin (17-AAG)-loaded poly(lactic acid-co-glycolic acid) nanoparticles. Archives of Pharmacal Research 38: 930-931.
- Hu YB, Dammer EB, Ren RJ, Wang G (2015) The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener 4.
- Abbey-Berko Y, Akala EO (2020) Computer Optimization of Stealth Biodegradable PolymericDual-Loaded Nanoparticles for Cancer Therapy using CentralComposite Face-Centered Design. Pharmaceutical Nanotechnology 8: 1-25.
- Ma Y, Zheng Y, Zeng X (2011) Novel docetaxel-loaded nanoparticles based on PCL-Tween 80 copolymer for cancer treatment. Int J Nanomed 6: 2679-2688.
- Mateja C, Janko K, Julijana K (2004) Cystatin incorporated in poly(lactide-co-glycolide) nanoparticles: development and fundamental studies on preservation of its activity. Euro J Pharma Sci 22: 357-364.
- Lundberg BB (2011) Preparation and characterization of polymeric pH-sensitive stealth nanoparticles for tumor delivery of a lipophilic prodrug of paclitaxel. International J Pharma 408: 208-212.
- Zhang Y, Chen Y, Westerhoff P, Crittenden J (2009) Impact of natural organic matter and divalent cations on the stability of aqueous nanoparticles. Water Res 43: 4249-4257.
- Patil S, Sandberg A, Heckert E, Self W, Seal S (2007) Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials 28: 4600-4607.
- Porter JR, Fritz CC, Depew KM (2010) Discovery and development of Hsp90 inhibitors: A promising pathway for cancer therapy. Current Opinion in Chemical Biology 14: 412-420.
- Chou T-C, Talalay P (1984) Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Advances in Enzyme Regulation 22: 27-55.
- Chou T-C (2006) Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacological Reviews 58: 621-681.
- Steel RG, Torrie HH (1980) Principles and Procedure of Statistics, A Biometrical Approach. McGraw-Hill Book Company NY (2nd eds).
- Foucquier J, Guedj M (2015) Analysis of Drug Combinations: Current Methodological Landscape. Pharmacology Research & Perspectives 3.
- Bhaskar S, Tian F, Stoeger T (2010) Multifunctional Nanocarriers for Diagnostics, Drug Delivery and Targeted Treatment across Blood-Brain Barrier: Perspectives on Tracking and Neuroimaging. Particle and Fibre Toxicology 7.
- Drbohlavova J, Chomoucka J, Adam V (2013) Nanocarriers for Anticancer Drugs.
- New Trends in Nanomedicine. Curr Drug Metab 14: 547-564.
- Su H, Wang Y, Gu Y, Bowman L, Zhao J, et al., (2018) Potential Applications and Human Biosafety of Nanomaterials Used in Nanomedicine. J Appl Toxicol 38: 3-24.
- Davis ME, Chen Z (2008) Nanoparticle Therapeutics: An Emerging Treatment Modalityfor Cancer. Nat Rev Drug Discov 7: 771-782.
- Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, et al., (2007) Combination oftrastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol 25: 5410-5417.
- Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nature (Cancer) 10: 537-549.