Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 7 Issue: 1
Nanoparticle Formulation of Brusatol: A Novel Therapeutic Option for Cancers
Adesina SK* and Reid TE
Department of Pharmaceutical Sciences, Howard University, Washington DC, USA
*Corresponding Author : Simeon K
Adesina, R.Ph., Ph.D, Department of Pharmaceutical Sciences, College of Pharmacy, Howard University, Washington, DC 20059, USA
Tel: 202-864-0401
Fax: 202-806-7805
E-mail: simeon.adesina@howard.edu
Received: January 18, 2018 Accepted: February 28, 2018 Published: March 07, 2018
Citation: Adesina SK, Reid TE (2018) Nanoparticle Formulation of Brusatol: A Novel Therapeutic Option for Cancers. J Pharm Drug Deliv Res 7:1. doi: 10.4172/2325-9604.1000174
Abstract
Objective: Challenges to the use of brusatol for cancer chemotherapy include its reversible and short-lived effect on Nrf2 which is limited to a few hours, its non-selective inhibition of protein synthesis which renders it potentially toxic to non-cancerous cells resulting in adverse effects and poor water solubility. A nanoparticle formulation of brusatol is expected to overcome these challenges and facilitate the clinical use of brusatol. In this proof-of-principle study, a brusatol-loaded nanoparticle formulation is developed and characterized.
Method: Brusatol-loaded mPEG-PLGA nanoparticles were prepared using the oil-in-water emulsification solvent diffusion method and characterized. The drug content of the nanoparticle formulation was determined by High Performance Liquid Chromatography. Toxicity of the brusatol-loaded nanoparticles in prostate cancer cell lines was evaluated over 120 hours using the Cell Titer 96® Non- Radioactive Cell Proliferation Assay and nanoparticle uptake was studied by confocal microscopy.
Results: Scanning electron microscopy revealed the formation of nanoparticles. The average hydrodynamic particle size is 309.23 ± 2.3 nm. The in vitro release isotherm showed a biphasic and sustained release of the encapsulated drug. Data from cytotoxicity studies reveal that the nanoparticle formulation showed more toxicity compared to control brusatol solution in PC-3 and LNCaP cell lines. Confocal microscopy studies showed internalization of the nanoparticles in PC-3 cells at 6 hours. In addition, z-stack images confirm the presence of nanoparticles at various depths within the cells.
Conclusion: The stealth nanoparticle formulation allows the sustained release of brusatol with the potential to modulate its short-lived effect on Nrf2. In addition, the potential of the nanoparticle formulation to target the tumor microenvironment via the enhanced permeability and retention effect and prevent toxicity to non cancerous cells is achieved. We report the preparation and characterization of a stealth nanoparticle formulation of brusatol to facilitate the clinical use of the drug for he treatment of cancers.
Keywords: Oxidative stress; Site-specific delivery; Cytotoxicity; Brusatol; Nuclear factor erythroid 2-related factor 2; Protein synthesis inhibitor; Nanoparticle; Sustained release
Introduction
Prostate Cancer (PC) is the second leading cause of cancer deaths and the second most frequently diagnosed cancer in men in the United States [1]. Androgen deprivation therapy eventually fails resulting in the presence of Androgen-Independent Prostate Cancer [AIPC] cells [2-4]. Once AIPC (also known as castrate-resistant prostate cancer (CRPC)) develops, tumor response to hormonal therapy and chemotherapy are of short duration with mean survival time of approximately 18 months [3]. Also, chemotherapeutic agents that are active against rapidly dividing cells may be relatively ineffective against metastatic AIPC because these cancers have a low proliferative rate [2,5]. Currently, only docetaxel and cabazitaxel are being used to treat CRPC with only modest improvements in overall survival and quality of life [4]. Docetaxel resistance is a major concern in cancer chemotherapy [6]; thus, novel treatment approaches that target tumor cells that are slowly proliferating and approaches that can cause reversal of chemoresistance are greatly desired in the treatment of metastatic PC.
Oxidative stress results from an imbalance in the production of Reactive Oxygen Species (ROS) and their elimination by cytoprotective mechanisms [7]. The role of oxidative stress in the initiation and progression of cancers is well reported [8,9]. Generally, appropriate levels of ROS favors cell proliferation and increase cell survival while high levels of ROS can cause cellular toxicity and apoptosis [10,11]. In addition, data have shown that cancer cells generate high levels of reactive oxygen species (ROS) and the degree of ROS production has been correlated with the aggressiveness of the tumor cells [12]. To counteract the effect of increased levels of ROS and therefore prevent cellular toxicity, cancer cells also express increased levels of antioxidant proteins [13,14]. Thus, to facilitate toxicity to cancer cells and promote cancer cell death, approaches to tilt the redox balance to favor increased ROS levels and facilitate cellular toxicity are appealing [7,13,15].
Numerous studies have shown that nuclear factor erythroid 2-related factor 2 (Nrf2), a redox sensitive transcription factor, is the master regulator of cellular defense mechanisms against xenobiotic and oxidative stress [14,16]. It is also involved in the induction of drug efflux pumps and metabolizing enzymes and thus plays an important role in drug response and chemoresistance [9,17]. Nrf2 has been shown to have a dual role in cancer. In the first case, Nrf2 activation under oxidative stress induces the transcription of about 100 cytoprotective genes [17]. In this regard, Nrf2 pathway activation in response to oxidative stress or xenobiotic insults, induces the expression of cellular protective genes that bear an antioxidant response element in their regulatory regions, thereby enhancing cell survival [18]. Increased expression of antioxidant molecules is an effective way to reduce ROS levels and allow cancer cells to survive under xenobiotic stress and ROS-mediated mutations [19]. Thus, therapy involving inhibition of the Nrf2 pathway and consequently the Nrf2-mediated cytoprotective mechanisms is a valid strategy for cancer treatment and to reverse acquired resistance. This strategy may present a paradigm shift in the treatment of cancers [9]. Secondly, recent findings suggest that Nrf2 promote cancers as a result of mutations that disrupt the negative regulation of Nrf2. This gives rise to high levels of Nrf2 in cancers and correlates with chemoresistance in cancer cells [16,18].
Brusatol (Figure 1), isolated from an evergreen shrub Brucea javanica (L) has been reported to reduce Nrf2 protein levels and inhibit Nrf2 signaling [9,16]. By this mechanism, it has been reported to facilitate tumor regression, and reduce chemoresistance in both In vitro and In vivo cancer models [16]. Previous data have also shown that brusatol inhibits eukaryotic protein synthesis [20,21]. Thus, brusatol is an excellent candidate for cancer therapy based on its reported mechanisms of action as a general protein synthesis inhibitor thereby facilitating nonspecific cytotoxicity to cells, and as a potent inhibitor of the Nrf2 pathway triggering ROS-induced cell death. In addition, brusatol has been reported to be effective in eradicating putative cancer stem cells and thus may be utilized in combination with other cytotoxic drugs to improve therapeutic efficacy [14] (Figure 1).

Figure 1: Chemical structure of brusatol.
Potential challenges to the clinical translation and development of brusatol in the treatment of cancer have been reported. These include its reversible and short-lived effect on Nrf2 which is limited to a few hours followed by a rebound of Nrf2 levels after treatment[[9,16]. However, data have shown that repeated administration of brusatol in mice correlated with greater antitumor efficacy [9]. Another potential challenge is related to its non-selective mechanism of action which renders it potentially toxic to non-cancerous cells. The non-selective inhibition of protein synthesis and Nrf2 may render non-target cells sensitive to the cytotoxic effects of brusatol and also to the effects of other cytotoxic drugs resulting in greater incidence of adverse effects [16]. Another potential challenge with the clinical translation of brusatol is poor bioavailability as a result of poor water solubility [22].
To facilitate the clinical use of brusatol in the treatment of cancers and other disease states, the fabrication and development of nanoparticle formulations of brusatol is expected to confer significant delivery advantages. First, the transient brusatol effect on Nrf2 can be reversed by a sustained, continuous release from a nanoparticle dosage form thereby ensuring sustained action on Nrf2. Second, delivery in a nanoparticle platform holds the potential to reduce adverse effects and increase therapeutic efficacy as a result of site-specific delivery via active and passive delivery mechanisms (targeting). By targeting, the drug is released at the site of action conferring selective toxicity to cancer cells without toxicity to neighboring healthy cells. Nanoparticle delivery also allows synchronous delivery of multiple drugs (combination therapy) which is essential to suppress chemoresistance and also improve therapeutic efficacy. Third, formulation in a nanoparticle dosage form also circumvents problems associated with poor aqueous solubility. In this paper, we report the preparation and characterization of brusatolloaded nanoparticles brusatol-loaded nanoparticles and its evaluation in vitro using prostate cancer cell lines.
Material and Methods
Materials
Methoxy-terminated polyethylene glycol - poly [lactide-coglycolide] [mPEG-PLGA] [lactic acid: glycolic acid 1:1] was purchased from Polyscitech® [Akina Inc., West Lafayette, IN, USA]. Polyvinyl alcohol [PVA 99-100%, MW 86,000] was purchased from ACROS Organics [Morris Plains, NJ, USA]. Brusatol was purchased from Carbosynth [San Diego, CA, USA]. All other solvents were purchased from Sigma-Aldrich [St. Louis, MO, USA].
Methods
Preparation and characterization of Brusatol-loaded PLGAPEG nanoparticles: The oil-in-water (o/w) emulsification solvent diffusion method was modified to prepare nanoparticles [23-25]. Using this method 50 mg of mPEG-PLGA was dissolved in dichloromethane. Brusatol was dissolved in a dimethyl sulfoxide: acetone (1:4) mixture and added to the PLGA solution to form the organic phase. The aqueous phase used was polyvinyl alcohol solution (1% w/v). High intensity probe sonication [Vibra-Cell; Model VC 750, Sonics and Materials, Newton, CT] was used to emulsify the organic phase in the aqueous phase followed by solvent evaporation to precipitate the particles. Nanoparticles were collected by centrifugation followed by washing with deionized water. The nanoparticle suspension was flashfrozen over liquid nitrogen and lyophilized for 48 hours. To prepare rhodamine-123 loaded nanoparticles, brusatol was replaced with rhodamine 123 in the formulation.
Particle size and size distribution analysis: Particle size and size distribution of nanoparticles was determined by Dynamic Light Scattering (DLS) using a 90 Plus particle size analyzer (Brookhaven Instruments Corp., NY, USA). Before analysis, the dilute nanoparticle suspension was filtered through a syringe filter with 5 μm pore size (Millex-SV, Merck Millipore, Cork, Ireland). Particle size was determined at 25ºC. The mean of three measurements was recorded. The particle size distribution is given by the Polydispersity Index (PDI).
Determination of drug content of nanoparticles: Drug loading, defined as the weight percent of drug in the nanoparticle formulation, was determined. The amount of brusatol in the nanoparticles was quantified by a HPLC method from standard calibration curves of pure drug. Briefly, a known amount of freeze-dried nanoparticles was dissolved in acetonitrile. The solution was then filtered through a 0.22 μm syringe filter and the amount of drug dissolved in the solution was quantified using a validated HPLC method on Agilent series 1100 HPLC equipped with a Zorbax Eclipse plus C18 column kept at 37ºC. The mobile phase for HPLC studies is 0.05% v/v phosphoric acid: acetonitrile (60:40) at a flow rate of 1 mL/min. Quantitation of brusatol was carried out using a diode array detector at 235 nm.
The percent drug loading for each drug was calculated from the equation below:
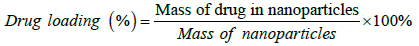
Scanning Electron Microscopy [SEM] of nanoparticles: The surface morphology of the nanoparticles was evaluated using a JSM- 7600F Scanning electron microscope (JEOL USA, Inc. Peabody, MA). To evaluate surface morphology, the nanoparticle suspension in distilled water was placed on a carbon tape affixed to a specimen stub and dried in vacuo. After drying, the samples were gold-coated prior to viewing. Images were taken at different sample magnifications.
Infrared spectroscopy studies: Infrared spectroscopy studies were carried out using a Spectrum 100 Fourier Transform Infrared (FTIR) spectrophotometer (Perkin Elmer, Shelton, CT, USA). FT-IR spectra were acquired for brusatol, brusatol-loaded nanoparticles and blank nanoparticles and overlaid.
Determination of brusatol release profile: The release profile was determined using a modified published method [26]. Briefly, brusatol-loaded nanoparticles were dispersed in 2 mL of Phosphate Buffered Saline [PBS] and placed in a dialysis bag (molecular weight cut off of 12,000-14,000). The dialysis bag was then immersed in a 15-mL Eppendorf tube containing a known amount of PBS. The tube was clamped to a LABQUAKE shaker rotated at 360° and maintained at 37°C. At different time intervals, an aliquot of the release medium was taken and replaced with fresh PBS to maintain sink conditions. The sample was diluted with acetonitrile and filtered through a 0.22 μm syringe filter and the amount of brusatol released into the solution was quantified by HPLC.
Cell cultures: The human prostate cancer cell lines, PC-3 and LNCaP, were obtained from American Type Culture Collection (Manassas, Virginia). PC-3 and LNCaP cells were maintained in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum and 100 U/mL of penicillin G and 100 g/mL of streptomycin sulfate. The cells were maintained as a monolayer in an incubator at 37°C in a humid atmosphere with 5% carbon dioxide.
Cytotoxicity studies: PC-3 (seeding density of 3,000 cells per well) and LNCaP (seeding density of 6,000 cells per well) were seeded in 96-well plates and allowed to attach for 24 h. To determine biocompatibility of blank nanoparticles, PC-3 cells were initially treated with culture medium containing blank nanoparticles of the same quantity expected to contain 10 nM, 50 nM, 100 nM and 500 nM concentrations of brusatol. To evaluate the cytotoxicity of brusatol-loaded nanoparticles, cells were treated with 100 μL of culture medium containing brusatol-loaded nanoparticles or brusatol in solution [to prepare the control brusatol solution, brusatol concentrations of 500 μM, 100 μM, 50 μM and 10 μM were prepared as stock solutions in sterile DMSO. Serial 1000-fold dilutions of these stock solutions were then prepared in RPMI to give growth media with drug concentrations of 500nM, 100 nM, 50 nM and 10 nM respectively each containing 0.1% DMSO. To allow direct comparison, the amount of brusatol-loaded nanoparticles containing the same amount of brusatol as the brusatol solution was used. Control cells were treated with culture medium only and culture medium with 0.1% DMSO. Cell viability was assessed at 120 h after treatment. The Cell Titer 96® Non-Radioactive Cell Proliferation Assay (Promega Corp.) was employed for this study. Results are presented as percent viability normalized to controls and represent the mean ± SD of six replicates per concentration tested. Results were analyzed using Oneway Analysis of Variance (ANOVA) and Student’s t-test with the aid of SPSS® statistical software. Differences were considered significant at p < 0.05.
Cell cycle experiments: LNCaP cells (seeding density of 40,000 cells per well) were seeded in triplicates in a 24-well plate and incubated for 48 hours. Cells were treated with 1 mL of 50 nM brusatol solution for 120 h and control cells were treated with 1 mL of culture media containing 0.01% DMSO. The experiment was done in triplicate. At the 120 h time point, cells were harvested and washed twice with cold PBS, fixed in 70% ethanol for 1 h at 4oC. The cells were then washed twice with PBS, and treated with ribonuclease followed by staining with propidium iodide. The stained cells were subjected to cell cycle analysis using the BD AccuriTM C6 flow cytometer (BD Biosciences, Ann Arbor, MI, USA). For each replicate, 20,000 events were recorded.
In vitro intracellular nanoparticle localization: Cellular uptake of fluorescent nanoparticles was investigated using spinning-disk confocal microscopy. PC-3 cells (10,000 cells/well) were seeded in a 24 well plate containing coverslips. After 24 hours, the media was aspirated and replaced with fresh media containing 50 μg/ml of rhodamine-123-labeled nanoparticles and incubated for 6 hours. The cells were washed 4 times with PBS to remove nanoparticles that were not internalized followed by membrane staining with CellMask Deep Red plasma membrane stain (4 μg/mL) for 15 min. The cells were washed twice with PBS and fixed with 4% paraformaldehyde for 20 min followed by DAPI (0.08 μg/mL) staining of chromosomal DNA for 10 min. Fluorescent images of fixed samples were captured using a Nikon Eclipse Ti microscope equipped with a 60x 1.4NA Plan Apo Lamda objective. Fluorescent signals of DAPI, rhodamine-123 and Cell Mask Deep Red were collected after excitation with 405-, 488- and 640 nm laser lines respectively. Images were acquired using NISElements software.
Results
Preliminary screening cytotoxicity studies
Preliminary cytotoxicity study carried out using a broad range of concentrations of brusatol solution on PC-3 and LNCaP cell lines over 72 hours revealed toxicity to both prostate cancer cell lines. Data revealed impressive killing of these cancer cells (Figure 2).

Figure 2: Cytotoxicity studies of different concentrations of brusatol relative to 0.1% DMSO control using PC-3 cells (Top) and LNCaP cells (bottom).
Preparation and characterization of nanoparticles
We have developed a solvent system that allows us to dissolve multiple drugs with varying solubility. We have used the solvent system to prepare blank and brusatol-loaded nanoparticles using the oil-in-water (O/W) emulsification solvent diffusion method in this work. Scanning electron microscopy revealed the formation of spherical nanoparticles (Figure 3).

Figure 3: Representative SEM micrograph of brusatol-loaded nanoparticles.
The hydrodynamic particle size was determined by DLS (Figure 4). The mean effective diameter obtained for the blank nanoparticle formulation is 267.8 ± 3.7 nm. After loading with brusatol, the mean effective diameter increased to 309.23 ± 2.3 nm. The polydispersity is an indication of the size distribution of nanoparticle formulations. Data obtained for blank and brusatolloaded nanoparticles show that the polydispersity is 0.143 ± 0.051 and 0.178 ± 0.098 respectively.

Figure 4: Particle size analysis data of brusatol-loaded nanoparticles by dynamic light scattering. The effective diameter recorded is 309 ± 2.3 nm with a polydispersity index of 0.178 ± 0.098.
The drug content of the nanoparticle formulation was determined by HPLC. The percent drug loading obtained was 1% w/w. FT-IR studies reveal that the drug loading determined was due to encapsulated drug and not due to drug adsorbing to the surface of the nanoparticles (Figure 5). The spectrum obtained for blank nanoparticles is similar to that for drug-loaded nanoparticles and no brusatol peak was visible in the spectrum of the drug-loaded nanoparticles.

Figure 5: Fourier transforms infrared spectroscopy data. (a) Blank nanoparticles (top), (b) brusatol-loaded nanoparticles (middle), (c) pure brusatol (bottom).
Release profile of Brusatol-loaded nanoparticles
The In vitro release of brusatol from brusatol-loaded PLGAPEG nanoparticles was evaluated by a dialysis method in phosphate buffered saline (PBS) maintained at 37°C in a laboratory oven using a Labquake shaker. The in vitro release isotherm show sustained release of the encapsulated drug over 866 hours (36 days) with less than 30% of the drug payload released in 24 hours and about 50% released in 144 hours (Figure 6). In addition, the release was biphasic with an initial burst release followed by a period of gradual release.

Figure 6: In vitro release isotherm for brusatol-loaded PLGA-PEG nanoparticles in PBS at 37°C. Standard deviation (1.46% - 8.13%; n = 3).
Cytotoxicity studies
The data obtained from evaluation of toxicity of the blank nanoparticle formulation in PC-3 cells show that the blank nanoparticles were not toxic at the highest concentration tested which is equivalent to the amount of polymer in 500 nM of brusatol-loaded nanoparticles (Figure 7). Percent viability values of greater than 95% was obtained for all the concentrations tested.

Figure 7: Evaluation of cytotoxicity of blank nanoparticles using the Cell Titer 96® Non-Radioactive Cell Proliferation Assay at 120 hours in PC-3 prostate cancer cells (n=6).
In addition, cytotoxicity data reveal that both brusatol-loaded nanoparticles and the control brusatol solutions at the same concentrations exhibited dose-dependent toxicity to PC-3 and LNCaP prostate cancer cell lines at the concentrations used in this study at 120 hours (Figure 8a, 8b). Generally, the nanoparticle formulation showed more toxicity to both cell lines compared to brusatol solution.

Figure 8: Evaluation of cytotoxicity of brusatol solution and brusatol-loaded nanoparticles using the Cell Titer 96® Non-Radioactive Cell Proliferation Assay at 120 hours. (a) PC-3 cells (top), (b) LNCaP cells (bottom) (n=6).
Cell cycle analysis
Cell cycle analysis of LNCaP cells treated with 50 nM of brusatol for 5 days revealed that the drug induced a G1 arrest, with considerably reduced number of cells progressing to the S and G2/M phases compared to control cells (Figure 9).

Figure 9: Cell cycle analysis. Treatment of LNCaP cells with brusatol (50 nM) for 120 h induces a G1 arrest. Control cells (green curve); brusatoltreated cells (red curve).
Nanoparticle uptake studies
Cellular uptake of fluorescent nanoparticles was evaluated using confocal microscopy after a 6-hour incubation of rhodamine-123- loaded nanoparticles with PC-3 cells. Our data show internalization of the nanoparticles at 6 hour. Figure 10 shows the rhodamine- 123-loaded nanoparticles (green color) around the nucleus (blue color) and enclosed by the cell membrane (red color). In addition, z-stacks images by confocal microscopy reveal the presence of nanoparticles at various depths within the cells which confirm that the nanoparticles were internalized by the cells and not adhering to the cells (supplementary data; Figure S1).

Figure 10: Uptake of fluorescent nanoparticles by PC-3 cells at 6 hours. Lower left quadrant shows rhodamine-123-loaded nanoparticles only; lower right quadrant shows cell membrane staining only; upper right quadrant shows nuclei staining only and upper left quadrant shows overlay of all the quadrants.
Discussion
With current therapy, prostate cancer eventually progresses to castration resistant prostate cancer, metastases and death [27] as a result of hormone refractory state due to the presence of androgenindependent prostate cancer cells [2]. These androgen-independent cells, generally considered to be androgen-independent Cancer Stem Cells [CSCs], have been isolated from clinical samples, xenograft tumors and cancer cell lines with evidence of CSC properties [28,29]. Thus, to eradicate tumors, newer therapies to which CSCs are sensitive must be employed in addition to other chemotherapeutic agents. Another approach is the tumor-specific delivery of cytotoxic drugs to limit toxicity to tumor cells. This approach will minimize the incidence of adverse effects in healthy non-cancerous cells and improve therapeutic efficacy. The use of a nanoparticle platform for targeting cancers as an example of this approach has several advantages. Nanoparticle drug delivery allows combination therapy, longer circulation times and controlled drug release.
Prostate cancer is a heterogeneous disease and combination therapy may be the best modality to treat advanced prostate cancer to enhance therapeutic efficacy and improve survival [30]. Brusatol however, has been reported to possess a unique potential to inhibit both putative cancer stem cells [14] and bulk tumor cells i.e. it can inhibit tumor cells and those cells that generate tumor cells. In addition, the drug can target two distinct pathways that are important in the progression of cancers as a result of its action on Nrf2 and as a global protein synthesis inhibitor [9,16,20,21]. These mechanisms make it potentially toxic to both healthy and tumor cells. In this proofof- concept paper, we report the preparation, characterization and in vitro testing of brusatol-loaded nanoparticles with the potential for site-specific delivery to improve efficacy and avoid toxicity.
Poly (lactide-co-glycolide) is approved by the US Food and Drug Administration for use in humans which makes its use for nanomedicines attractive [31]. PLGA-PEG was used in this work to facilitate prolonged circulation in blood when administered in vivo. Brusatol-loaded nanoparticles were prepared by the oil-in-water (o/w) emulsification solvent diffusion method. This method has been reported to be good for loading hydrophobic drugs such as brusatol into PLGA nanoparticles [32]. Scanning electron microscopy reveals the formation of smooth spherical nanoparticles (Figure 3). Particle size analysis by dynamic light scattering reveal that the nanoparticle size increased from 267.8 ± 3.7 nm to 309.23 ± 2.3 nm after drug loading. Numerous studies have reported an increase in particle size with increase in drug content. The increase in size may be ascribed to an increase in amount of drug present in emulsion nanodroplets during nanoparticle preparation [33,34].
The drug loading of brusatol also referred to as drug content was determined by HPLC from standard calibration curves of pure drug. The drug loading was determined to be 1% w/w. Optimization of drug loading is currently ongoing and was not carried out in the present study. The in vitro release profile of brusatol was determined in PBS (pH 7.4). Data show an initial burst release of the encapsulated brusatol followed by a more sustained release over 36 days (Figure 6). It has been reported that polymer coating of a nanoparticle acts as a drug release barrier and consequently impacts drug release [35]. Generally, the burst release is attributed to dissolution and the subsequent diffusion of poorly entrapped drug in the polymer matrix while the more sustained release is governed by diffusion and matrix erosion from the PLGA core [26,35-37]. The observed sustained release of brusatol from the nanoparticle is advantageous to ensure a sustained action on Nrf2 and thus counteract its reported transient effect. In addition, FT-IR data (Figure 5) does not reveal the presence of surface drug, thus, the calculated drug loading and the burst effect observed is clearly not due to the presence of surface drug molecules but encapsulated drug.
The toxicity of the brusatol-loaded nanoparticles in prostate cancer cell lines was evaluated over 120 hours using the Cell Titer 96® Non-Radioactive Cell Proliferation Assay (Promega Corp.). The characteristics of the cell lines utilized in this study are well known. LNCap cells are androgen receptor dependent while PC-3 cells are androgen independent [38]. This allows us to evaluate the effect of brusatol on both androgen dependent and androgen independent cells. Our data shows that blank nanoparticles are biocompatible at the highest concentration tested which is equivalent to the amount of polymer in 500 nM of brusatol-loaded nanoparticles. Greater than 95% viability was observed in both cell lines after treatment with blank nanoparticles for 120 h. The data also confirm that the cytotoxicity observed with brusatol-loaded nanoparticles is due to the release of the encapsulated drug from the nanoparticle delivery system. In addition, the cytotoxicity observed could not be attributed to androgen sensitivity as the brusatol nanoparticle formulation was revealed to be toxic to both PC-3 (IC50 of 34 nM) and LNCaP (IC50 of 16 nM) cell lines. This confirms its reported mechanism of action as a global inhibitor of protein synthesis and its effect on Nrf2 which are not correlated to androgen sensitivity. Furthermore, cell cycle analysis of LNCaP cells treated with brusatol solution (50 nM) reveals a G1 arrest (Figure 9). This is consistent with prior data on the effect of brusatol in leukemia cells [39]. G1 arrest is associated with inhibition of protein synthesis [40,41] in agreement with its reported mechanism of action as a protein synthesis inhibitor [20,21].
Statistical analysis of cytotoxicity data reveals a significant difference between the drug in solution and the nanoparticle formulation of the drug. At test concentrations close to the IC50, the nanoparticle formulations containing brusatol were significantly more toxic to cells compared to the solution of the drug. Post hoc tests (Bonferroni) consequent to one-way analysis of variance (ANOVA) reveal that for PC-3 cells, there was a significant difference in viability between the nanoparticle formulation and the drug in solution at the 50 nM concentration tested at 5% level of significance (p<0.05). For the LNCaP cell line, there was a significant difference in viability between the nanoparticle formulation and the drug in solution at both 10 nM (p<0.05) and 50 nM (p<0.05) concentrations at 5% level of significance. At higher concentrations, there was no significant difference in toxicity between the nanoparticle formulation and the drug solution. This may be adduced to the fact that at high concentrations, both formulations were considerably toxic to cells such that any difference in toxicity between the formulations is obscured. Furthermore, statistical analysis of IC50 data using the T test reveal that LNCap cells are significantly more sensitive to brusatol compared to PC-3 cells at 5% level of significance (p<0.05).
In vitro drug release studies show that less than 50% of brusatol was released at 120 hours. Data from cytotoxicity studies reveal that the nanoparticle formulation was more toxic to both cancer cell lines when compared to the drug in solution formulation. The question then arises - why is the nanoparticle formulation more effective in killing cells despite the fact that the solution dosage form has a higher concentration of drug available to cells? To answer the question, confocal microscopy studies to evaluate nanoparticle cellular internalization was conducted to determine the mechanism of greater cytotoxicity of the nanoparticle formulation. Data show intense uptake of nanoparticles into the cell cytoplasm at the 6 hour time point evaluated. Thus, drug release from nanoparticles occurs in the cell cytoplasm where the nanoparticles act as intracellular drug depots by slowly releasing the encapsulated drug. This potentially leads to a sustained inhibitory effect on Nrf2 and sustained inhibition of protein synthesis in the cytoplasm thereby leading to an increase in cytotoxicity of the nanoparticle formulation.
We are aware of reports that brusatol is not cytotoxic at 40 nM which is greater than the IC50 values determined in our study [14]. These reports were based on studies conducted on tumor cell lines different from our own. The difference in these studies may be a consequence of differences in the cell lines used or as a result of the duration of our experiment (120 h). Previous data has shown that the duration of cytotoxicity studies has considerable effect on cytotoxicity of drugs [37]. However, our choice of duration is apt as drug-loaded nanoparticles are designed to be retained at tumor sites in vivo by the EPR effect and release the drug payload continuously over a prolonged period of time.
Conclusions
Brusatol-loaded nanoparticles have been successfully prepared and characterized in this study. The nanoparticles were tested on prostate cancer cell lines in vitro. Data show that the free drug in solution and the nanoparticle formulation of the drug inhibited the growth of both PC-3 and LNCaP prostate cancer cells. However, the nanoparticle formulation showed significantly higher cytotoxicity compared to the free drug in solution at lower concentrations. Further optimization studies are required to reduce particle size and improve drug loading. We have established the proof of concept that the site-specific delivery of brusatol as exemplified by the brusatol nanoparticle formulation holds the potential to facilitate the clinical translation of the drug for the treatment of cancers by improving therapeutic efficacy, reversal of chemoresistance and reduction of adverse effects.
Acknowledgements
We would like to thank Dr. Zebalda Bamji for assistance with interpreting flow cytometry data. Special thanks to Dr. Emmanuel Akala for access to facilities of the Center for Drug Research and Development (CDRD), Howard University.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- American Cancer Society. Cancer Facts & Figures (2016). Atlanta: American Cancer Society.
- Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, et al. (2003) Prostate-Specific Antigen-activated Thapsigargin Prodrug as Targeted Therapy for Prostate Cancer. J Natl Cancer Inst 95: 990-1000.
- Karantanos T, Corn PG, Thompson TC (2013) Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 32: 5501-5511.
- Guo Y, Lin Z, Wang M, Dong Y, Niu H, et al. (2016) Jungermannenone A and B induce ROS- and cell cycle-dependent apoptosis in prostate cancer cells in vitro. Acta Pharmacol Sin 37: 814-824.
- Denmeade SR, Lou W, Lovgren J, Malm J, Lilja H, et al. (1997) Specific and Efficient Peptide Substrates for Assaying the Proteolytic Activity of Prostate-specific Antigen. Cancer Res 57: 4924-4930.
- Nagesh P, Johnson N, Boya V, Chowdhury P, Othman S, et al. (2016) PSMA targeted docetaxel-loaded superparamagnetic iron oxide nanoparticles for prostate cancer. Colloids Surf B Biointerfaces 144: 8-20.
- Nogueira V, Hay N (2013) Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implication for cancer therapy. Clin Cancer Res 19: 4309-4314.
- Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44: 1-31.
- Ren D, Villenueve NF, Jiang T, Wu T, Lau A (2011) Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci 108: 1433-1438.
- Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579-591.
- Ding S, Li C, Cheng N, Cui X, Xu X, et al. (2015) Redox regulation in cancer stem cells. Oxid Med Cell Longev Article ID 750798: 1-11.
- Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK (2008) Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 68: 1777-1785.
- Gibellini L, Pinti M, Nasi M, De Biasi S, Roat E, et AL. (2010) Interfering with ROS metabolism in cancer cells: The potential role of Quercetin. Cancers 2: 1288-1311.
- Wu T, Harder BG, Wong PK, Lang JE, Zhang DD (2015) Oxidative stress, Mammospheres and Nrf2-New Implication for Breast Cancer Therapy. Mol Carcinog 54: 1494-1502.
- Sullivan LB, Chandel NS (2014) Mitochondrial reactive oxygen species and cancer. Cancer Metab 2: 1-12.
- Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, et al. (2015) Brustaol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med 78: 202-212.
- Hayes JD, McMahon M (2008) NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34: 176-188.
- Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, et al. (2014) Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res 74: 7430-7441.
- Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR (2011) Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem 286: 37813-37829.
- Willingham Jr. W, Stafford EA, Reynolds SH, Chaney SG, Lee KH, et al. (1981) Mechanism of Eukaryotic Protein Synthesis Inhibition by Brusatol. Biochim Biophys Acta 654: 169-174.
- Vartanian S, Ma TP, Lee J, Haverty PM, Kirkpatrick DS, Yu K, Stokoe D (2016) Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis. Mol Cell Proteomics 15: 1220-1231.
- Chen M, Chen R, Wang S, Tan W, Hu Y, et al. (2013) Chemical components, pharmacological properties, and nanoparticles delivery systems of Brucea javanica. Int J Nanomedicine 8: 85-92.
- Niwa T, Takeuchi H, Hino T, Kunou N, Kawashima Y (1993) Preparations of biodegradable nanospheres of water-soluble and insoluble drugs with D, L-lactide/glycolide copolymer by a novel spontaneous emulsification solvent diffusion method, and the drug release behavior. J Control Release 25: 89-98.
- Esmaeili F, Ghahremani MH, Ostad SN, Atyabi F, Seyedabadi M, et al. (2008) Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J Drug Target 16: 415-423.
- Song X, Zhao Y, Wu W, Bi Y, Cai Z, et al. (2008) PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: Systematic study of particle size and drug entrapment efficiency. Int J Pharm 350: 320-329.
- Ogunwuyi O, Adesina SK, Akala EO (2015) D-Optimal mixture experimental design for stealth biodegradable crosslinked docetaxel-loaded poly-ÆÂ-caprolactone nanoparticles manufactured by dispersion polymerization. Pharmazie 70: 165-176.
- Shtivelman E, Beer TM, Evans CP (2014) Molecular Pathways and Targets in Prostate cancer. Oncotarget 5: 7217-7259.
- Zhou Y, Yang J, Kopecek J (2012) Selective Inhibitory effect of HPMA copolymer-cyclopamine conjugate on prostate cancer stem cells. Biomaterials 33: 1863-1872.
- Singh S, Chikara D, Mehrazin R, Behrman SW, Wake RW, et al. (2012) Chemoresistance in Prostate Cancer Cells Is Regulated by miRNAs and Hedgehog Pathway. PLoS One 7: e40021.
- Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, et al. (2015) Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med 373: 737-746.
- Kumari A, Yadav SK, Yadav SC (2010) Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 75: 1-18.
- Sah E, Sah H (2015) Recent trends in preparation of poly[lactide-co-glycolide] nanoparticles by mixing polymeric organic solution with antisolvent. J Nanomater Article ID 794601
- Krishnamachari Y, Madan P, Lin S (2007) Development of pH-and time-dependent oral microparticles to optimize budesonide delivery to ileum and colon. Int J Pharm 338: 238-247.
- Sharma N, Madan P, Lin S (2015) Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J Pharm Sci 11: 404-416.
- Singh R, Lillard JW (2009) Nanoparticle-based targeted drug delivery. Exp Mol Pathol 86: 215-223.
- Danhier F, Lecouturier N, Vroman B, Christine J, Marchard-Brynaert J, et al. (2009) Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J Control Release 133: 11-17.
- Adesina SK, Holly A, Kramer-Marek G, Capala J, Akala EO (2014) Polylactide-based Paclitaxel-loaded Nanoparticles Fabricated by Dispersion Polymerization: Characterization, Evaluation in Cancer Cell Lines, and Preliminary Biodistribution Studies. J Pharm Sci 103: 2546-255.
- Majumder PK, Sellers WR (2005) Akt-Regulated Pathways in Prostate Cancer. Oncogene 24: 7465-7474.
- Mata-Greenwood E, Cuendet M, Sher D, Gustin D, Stock W, et al. (2002) Brusatol-mediated induction of leukemic cell differentiation and G1 arrest is associated with down-regulation of c-myc. Leukemia 16: 2275-2284.
- Van den Bogert C, van Kernebeek G, Leij LD, Kroon AM (1986) Inhibition of mitochondrial protein synthesis leads to proliferation arrest in the G1-phase of the cell cycle. Cancer Lett 32: 41-51.
- White-Gilbertson S, Kurtz DT, Voelkel-Johnson C (2009) The role of protein synthesis in cell cycling and cancer. Mol Oncol 3: 402-408.