Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 11 Issue: 1
Tunable Dual-phase Dual-drug Delivery System using a PLGA Micro particle/PVA Hydrogel Composite
Timothy Eck*, Lucinda Lau, Ryan Bach, Erik Brewer
Department of Engineering, Rowan University, Glassboro, USA
Corresponding Author:Timothy Eck
Department of Engineering, Rowan University, Glassboro, USA
E-mail:ecktim73@students.rowan.edu
Received date: 10 December, 2021, Manuscript No. JPDDR-21-39156;
Editor assigned date: 14 December, 2021, Pre QC No. JPDDR-21-39156 (PQ);
Reviewed date: 28 December, 2021, QC No. JPDDR-21-39156;
Revised date: 08 December, 2021, Manuscript No. JPDDR-21-39156 (R);
Published date: 17 January, 2022, DOI:10.4172/2325-9604.1000203
Citation: Eck T et al.(202 2) Tunable Dual-phase Dual-drug Delivery System using a PLGA Micro particle/PVA Hydrogel Composite. J Pharm Drug Deliv Res 11:1.
Abstract
Current Drug Delivery Systems (DDSs) have demonstrated controlled long-term, sustained release but have only tried to diminish burst release. For applications like wound healing, there is a need for a DDS which is adjustable in both short- and long-term release, independent of each other. We present a tunable dual-phase dual-drug delivery system composed of drug-loaded polymer micro particles and drug-loaded hydrogel in this study. This system was created using lidocaine and
dexamethasone encapsulated in poly (D,L-lactide-co-glycolide) particles combined with lidocaine- and dexamethasone-loaded poly(vinyl alcohol) hydrogel. Hydrogel drug concentration and micro particle mass fraction were investigated for their impact on short-term and long-term release, respectively. A two weeklong
drug release study was performed with formulations varying only hydrogel drug concentration and micro particle mass fraction. Analysis of drug release revealed that hydrogel drug concentration individually alters short-term release and particle mass fraction individually alters long-term release.
Keywords: Drug release; PLGA; Micro particle; PVA; Hydrogel; Dual; Multiphase composite
Introduction
Achieving zero-order release, or those that follow “near zero-order”, is an ideal many drug delivery systems hope to attain [1]. Benefits of zero-order, or sustained, release profiles offer include maintaining constant therapeutic blood levels of a drug for desired periods [2], eliminating toxic peak and inadequate valley drug concentrations stemming from multiple bolus deliveries [3], increasing the bioavailability of therapeutic [4], improving therapeutics cost-benefit [5], and improving patient-compliance to prescribed dosages [6]. Multiple strategies have been employed to achieve this release profile, including biphasic polymer hydrogels [7], three-layer asymmetric floatable systems [8], double-layered porous films [9], and hydrogels with entrapped PLGA microspheres [10], among many others. Despite extensive advances in novel delivery methods, these predictable release patterns are often difficult to achieve in practice [11].
One frequent phenomenon and problem that has hindered those targeting controlled release methods are short-term or “burst” releases, characterized by an initial large bolus of drug that is eluted before the release rate reaches stable profile upon placement in the surrounding medium [12]. Burst release is typically regarded as a negative consequence of creating drug delivery devices [13], often leading to local or system toxicity, shorter half-life of drugs, therapeutically-inefficient systems, and more frequent dosing. Numerous attempts at mitigating burst release have been made when creating a Drug Delivery System (DDS) with limited success [8,10,14-17]. Other attempts aimed to explain and model the release patterns to make them more predictable [18-20]. Even with these efforts, controlling burst release profiles remains a challenge. Patients with BPPV frequently have physical, functional, and emotional difficulties that can affect their social and family lives. Their distress can have a negative impact on their Health-Related Quality of Life (HRQOL).
In contrast, some researchers have used this phenomenon to their advantage. Huang et al. [12] described favorable burst release conditions, including wound treatment applications, encapsulation of flavors, triggered burst released in targeted systems, and pulsatile release systems Lischer et al. [21] utilized anti-bacterial-containing plasma polymer coatings with burst releases for preventing the onset of bacterial colonization similarly, Setterstrom et al. [22] developed burst releasing antibiotic coatings for topical administration to wounds. While these groups acknowledge the favorable results of rapid release caused by this effect, little research exists on drug delivery systems that actively incorporate this effect, let alone optimize it.
Certain applications of drug delivery have demonstrated that using two different rates of release can be most beneficial for administering the drug [23]. A study by Kastellorizios [24] has shown, for instance, that to better combat the foreign body response against implantable devices, a large burst release of anti-inflammatory dexamethasone (100 ug) immediately after subcutaneous implant insertion followed by smaller, continuous doses each day (10 ug) provide optimal results. This investigation suggests that there is a valuable use for drug delivery systems which can integrate a burst release phase and zero-order release phase, both of which can be controlled independently.
In the work subject of this paper, we describe a dual-phase drug delivery system that controls both the burst, which we will refer to as fast-action, and long-term release phases separately and optimizes them in a sequence ideally suited for fighting inflammation, using dexamethasone and lidocaine simultaneously as model drugs. Particles were first created then characterized by their size, loading and encapsulation efficiency. They were then mixed into a hydrogel matrix to create a composite. To modify and test varying fast-action release profiles, raw Active Pharmaceutical Ingredient (API) was added directly into the hydrogel matrix. To test sustained release profiles, the fraction of particle mass was altered. The resulting composites were formed into cylindrical molds. These molds were used in a two week long release study. High Performance Liquid Chromatography (HPLC) analysis was used to determine the amount of drug released at each time point sampled. The drug release data demonstrated how raw API in the hydrogel and particle mass fraction shape the fast-action release (drug released up to 24 hours) and sustained release (drug released after 24 hours), respectively.
Experimental
Materials
Lidocaine and Mowiol® (poly (vinyl alcohol)) (Mw~145,000 Da) were purchased from Sigma Aldrich (St. Louis, MO). Dexamethasone was purchased from Alfa Aesar (Tewksbury, MA). Poly (D,L-lactide-co-glycolide) (PLGA) (50% ratio of lactic acid and glycolic acid, 4A) was purchased from Lakeshore biomaterials (Birmingham, AL). Dichloromethane was purchased from VWR BDH chemicals (Radnor, PA). HPLC grade acetonitrile was purchased from Honeywell (Mexico City, Mexico). Phosphate buffer solution, HPLC-grade acetonitrile, and HPLC-grade Tri Fluoracetic Acid (TFA) was purchased from Sigma Aldrich (St. Louis, MO). Trifluoro acetic acid was purchased from Thermo scientific (Rockford, IL). Deionized water was obtained from an EMD Millipore Milli-Q system (Darmstadt, Germany).
Micro particle fabrication
Single emulsion followed method described by Rawat [25]. Briefly, dexamethasone and lidocaine loaded PLGA microspheres were prepared by oil-in-water (o/w) emulsion solvent extraction/evaporation technique [26]. 3.375 g of PLGA was dissolved in 45 mL dichloromethane. 33.75 mg dexamethasone and 1125 mg were dissolved in the PLGA solution. This solution was then added slowly to 225 ml of a 2% (w/v) aqueous poly (vinyl alcohol) (PVA) and stirred at 1500 rpm for 2 min using an IKA RW 20 digital overhead mixer. This emulsion was stirred at 300 rpm under vacuum for overnight at 25°C. The resulting microspheres were washed three times with de-ionized water and vacuum dried for at least 48 hours.
Particle Size Analysis
The size distribution and morphology of the micro particles were determined using a scanning electron microscope. Images were taken using an FEI Quanta 600 ESEM (Hillsboro, OR) at the singh center at the University of Pennsylvania. ImageJ (National institutes of health, Bethesda, MD) software was used to measure the average diameter of particles. The scale was set using the scale bar from the image. Manual measurements were taken using ImageJ. Average diameter and standard deviation were reported from ImageJ.
Loading and Encapsulation Efficiency
An aliquot of particles was earmarked to determine loading and encapsulation efficiency. A small, measurable amount (at least 3 mg) of particles was dissolved in 1.5 mL of acetonitrile. The particle solution was analyzed via HPLC (method described below). Drug loading was calculated using equation 1:
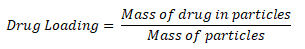
Encapsulation efficiency was calculated using Equation 2:
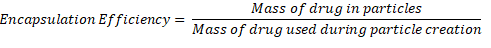
HPLC Method
A Waters (Milford, MA) High-Performance Liquid Chromatography (HPLC) system, equipped with a symmetry C18 column (150 mm, 5 µm, 100 Å) and Photodiode Array (PDA) UV/Vis detector and was used. Dexamethasone and lidocaine detection methods were guided by independent methods developed by Wang et al. [27,28]. Briefly, the mobile phases are HPLC grade water and acetonitrile with 0.1% TFA. We used a gradient method to obtain the dexamethasone and lidocaine concentration during one sampling cycle. The HPLC method starts with 90:10 ratio of water to acetonitrile for 8 minutes. The ratio then changes to 65:35 water to acetonitrile for 3 minutes. It then returns to 90:10 for 4 minutes which ends the sample analysis. The total run time is 15 minutes. UV/Vis detector collected data from 215 nm wavelength channel for lidocaine and 240 nm wavelength channel for dexamethasone.
Composite mold fabrication
PVA pellets were mixed into DI water at a ratio of 20% PVA: 80% water by weight. A stir bar was also placed in the container. The mixture was put into an autoclave at 121°C for 1 hour. The mixture was placed on a stir plate and mixed to homogeneity.
It was decided that 1 g of total composite would be used for each formulation mold. Four formulations were made with varying amounts of free API and particles in the hydrogel matrix: NoAPI, WeakAPI, StrongAPI, and HalfPart. NoAPI was composed of 20% particles/80% PVA by mass with no further addition of API. WeakAPI was composed of 20% particles/80% PVA and supplemented raw API of lidocaine and dexamethasone equivalent to 1 times the drug mass in the particles (based on loading data). StrongAPI was composed of 20% particles/80% PVA and supplemented raw API of lidocaine and dexamethasone equivalent to 5 times the drug mass in the particles (based on loading data). HalfPart was composed of 10% particles/90% PVA and supplemented raw API of lidocaine and dexamethasone equivalent to the drug mass in WeakAPI (based on loading data).
To create these formulations, heated PVA gel was placed into syringes in combination with prepared micro particles and raw API, if called for. All components were mechanically mixed to homogeneity and transferred via syringe into a 1 mL transfer pipette. The molded samples then underwent freeze-thaw cycles of 4hrs thaw/20hrs freezing, as described by Wang [27], to solidify the gels. Once solidified, the molds were divided into 3 pieces and weighed in preparation for release studies.
Release studies
Drug release kinetics were evaluated for all formulations by submerging the formulation mold, approximately 300mg each, in 4 ml of 37°C PBS, weighed gravimetrically. 500 uL of release media was taken at various time points (1 hour, 6 hours, 24 hours and every 24 hours hence), and the samples were replenished with equal volumes of fresh media. Samples were frozen until tested for concentration.
Analysis of HPLC data
Using dexamethasone and lidocaine standards, the concentration of each sample was determined using a gradient HPLC method. Each sample was compared to the standards curve. From this comparison, the drug released over time could be determined.
Using the concentration data obtained from the HPLC, the release of drug versus time was presented in several manners. For each formulation, a sample size of 3 was used. The mass of drug released per mass of particles was charted over the course of the release study.
Results and Discussions
Particle size analysis
An analysis of 770 particles revealed an average diameter of 18.6 um ± 11.2 um with a range of 2.9 um-94.0 um, indicating a large polydispersity. The histogram below shows the frequency distribution of particle diameters. The micro particles are spherical and have a smooth surface, indicative that there is negligible API present on their surface. There is some material which does not appear to be particles. Energy dispersive X-ray spectroscopy determined material is not lidocaine or dexamethasone, implying it is likely PLGA as shown in Figure 1 (Figure 1).

Figure 1: SEM images of PLGA micro particles loaded with lidocaine and dexamethasone.
HPLC quantification
In Figure 2 shows an example chromatogram of lidocaine and dexamethasone as detected with the described gradient method. The first peak, which occurs around 7.8 minutes, is where lidocaine is detected. Lidocaine produces a stronger peak when detected by the 215 nm wavelength channel. The second peak, which occurs around 11.2 minutes, is where dexamethasone is detected. Dexamethasone produces a stronger peak when detected by the 240 nm wavelength channel (Figure 2).

Figure 2: Chromatogram of gradient method separating dexamethasone and lidocaine.
Drug loading and efficiency
Drug loading in the particles used for this study were as follows: lidocaine=11.2% and dexamethasone=0.398%. Encapsulation efficiency in the particles used for this study was as follows: lidocaine=15.3% and dexamethasone=17.8%.
Release studies
Figure 3 shows the release of lidocaine and dexamethasone normalized per mass of particles comparing NoAPI, WeakAPI and StrongAPI over the course of 13 days (Figure 3).

Figure 3: Normalized drug release profile of lidocaine (left) and dexamethasone (right) from no API, weak API and strong API formulations (n=3).
In the first 24 hours, all formulations exhibited fast-action release profiles: the NoAPI, WeakAPI, and StrongAPI formulations released 77 ug, 118 ug and 220 ug of lidocaine/mg of particles, respectively, while also releasing 1.4, 2.7 and 5.8 ug of dexamethasone/mg of particles, respectively (Figure 3). Furthermore, all formulations demonstrated sustained, controlled release profiles at a slower rate than fast-action: After 24 hours through 13 days, the no API, weak API, and strong API formulations released 29 ug, 57 ug, and 43 ug of lidocaine/mg of particles, respectively, while also releasing 1.2 ug, 3.2 ug, and 2.9 ug of dexamethasone/mg of particles, respectively. Strong API, which contains the largest amount of drug mass in the hydrogel matrix, has the highest fast-action release, followed by weak API, which has an intermediate hydrogel drug mass, then no API, which has no hydrogel drug mass. This holds true for both lidocaine and dexamethasone.
Figure 4 shows the release of lidocaine and dexamethasone normalized per mass of particles comparing weak API and half part.

Figure 4: Normalized drug release profile of lidocaine (left) and dexamethasone (right) from weak API and half part.
In the first 24 hours, both formulations exhibited fast-action release profiles: The weak API and half part formulations released 118 ug and 207 ug of lidocaine/mg of particles, respectively, while also releasing 2.7 and 3.5 ug of dexamethasone/mg of particles, respectively (Figure 4). Furthermore, all formulations demonstrated sustained, controlled release profiles at a slower rate. After 24 hours through 13 days, the weak API and half part formulations released 57 ug and 116 ug of lidocaine/mg of particles, respectively, while also releasing 3.2 ug and 2.8 ug of dexamethasone/mg of particles, respectively. Weak API, which contains twice the mass of particles in the formulation, had a similar fast-action and sustained release of dexamethasone as half part. Half part had a higher fast-action and sustained release of lidocaine.
During fast-action release, the weak API and half part formulations released 67.4% and 62.5% of maximum lidocaine detected, totaling 7798 ug and 6174 ug, respectively, while also releasing 46.0% and 54.6% of maximum dexamethasone detected, totaling 181 ug and 105 ug, respectively. During sustained release, the weak API and half Part formulations released 31.7% and 36.1% of maximum lidocaine detected, totaling 3790 ug and 3545 ug, respectively, while also releasing 53.9% and 44.3% of maximum dexamethasone detected, totaling 213 ug and 84 ug, respectively. Weak API and half part showed similar percent release during fast-action and sustained release for both lidocaine and dexamethasone. WeakAPI released more overall lidocaine and dexamethasone during fast-action and sustained release than half part. This data is not shown in this paper.
PLGA particles and PVA hydrogel composites have been used often as successful methods of sustained drug release, achieving drug release on the scale of weeks [27-29]. These systems also typically exhibit an involuntary burst release. While some research has tried to mitigate burst release, there are benefits to incorporating burst release into drug delivery [12]. In this study, our team aimed to create such a composite which allowed for independent tuning of fast-action release and sustained release. Moreover, we aimed to do so using two model drugs simultaneously, with both drugs having a fast-action release phase followed by a sustained release phase. In our studies, we successfully engineered a composite that delivered a controlled fast-action release followed by a 13-day sustained release, concentrated in the first 4 days, using particles with an average diameter of 18.6 um ± 11.2 um.
Over the course of these experiments, our team investigated two hypotheses. We wanted to evaluate how free drug in the hydrogel matrix influences the fast-action release rate and how particle mass fraction influences sustained release. To test the first hypothesis, we created three formulations of composite with varying amounts of raw API present in the hydrogel matrix, measured as a ratio to particle-encapsulated drug. To test the second hypothesis, we created a fourth formulation which differed only in particle mass fraction compared to one of the first three formulations. The outputs we considered were mass of each drug released, mass of drug released per mass of particles, and mass of drug released relative to maximum drug release detected. We evaluated these outputs over the course of fast-action release (within 24 hours) and sustained release (after 24 hours until end of study).
In previous research, investigators have observed burst release by particle-based systems and tried to diminish the contribution due to burst. In this work, not only did we attempt to utilize this effect, we attempt to control it by increasing raw API content of the hydrogel portion of our system, relying on quicker dissolution kinetics relative to the particle release kinetics. We refer to this type of release as fast-action release. By comparing similar formulations that differ only their drug content in the hydrogel, we investigated how this increases the fast-action release. In Figure 3, we see the results of this study: In the formulations displayed, three different ratios of initial hydrogel drug mass to encapsulated drug mass were used. In all formulations, fast-action release was demonstrated to be significant and positively correlated with the addition of free API. Figure 3 shows that StrongAPI has the largest drug release within 24 hours, followed by weak API and then no API. This data supports the hypothesis that the amount of drug initially dissolved in the hydrogel correlates positively with the fast-action release. Comparing strong API to weak API, the drug released per mg of particles during fast-action release (within 24 hours) increases by a factor of 1.9 (lidocaine) and 2.1 (dexamethasone). With both drugs, more raw API added into the hydrogel increased fast-action release rate. However, we did not see the same effect on sustained release. Drug released per mg of particles during sustained release (after 24 hours) decreases by a factor of 0.75 (lidocaine) and 0.91 (dexamethasone). It is important to note that the increase observed in fast-action release was not present in sustained release.
The results of no API were a unique case. While both weak API and strong API showed the expected increase in fast-action release, both formulations also showed a noticeable increase in sustained release as well. This was not expected as all formulations contained comparable particle mass fractions. Comparing weak API to no API, the drug released per mg of particles during fast-action release (within 24 hours) increases by a factor of 1.5 (lidocaine) and 1.9 (dexamethasone) while drug released per mg of particles during sustained release (after 24 hours) increased by a factor of 1.9 (lidocaine) and 2.7(dexamethasone). We suspect this may be due to dissolution kinetics of the raw API lasting beyond the 24 hour period, a time generally recognized as the threshold of burst release [30]. In the future, we would exhibit further command over this factor by changing the size of raw API crystals. Several studies have established how the size of crystals impacts dissolution kinetics and how crystal size can be modified [31,32].
The fourth formulation, HalfPart, shares the same mass of free API in the hydrogel as the weak API formulation while it contains half of the PLGA particle mass fraction– weak API is 20% particles by mass, half part is 10%. Comparing these two formulations, we wanted to see how a decrease in particles affected the amount of drug eluted during the sustained release period. In the first 24 hours, half part releases 1.8 times more mg lidocaine per mg of particles and 1.3 times more mg dexamethasone per mg particles. When normalized with respect to the mass of particles, we would expect to see half part have twice as much drug release per mg of particles. The experimental results for lidocaine were reasonably close to this expectation. However, this was not the case with dexamethasone. We suspect that this could be due to measurement limitations. The very small amounts of dexamethasone required for these formulations were potentially beyond the required sensitivity of the available equipment leading to measurement errors. We expected that both formulations would release the same overall mass of drug during fast-action release as they contain equal amounts of drug mass in the hydrogel. The experimental results generally support this theory. There is a slight increase in this aspect for weak API over half part. This can be attributed to the fact that weak API contains more particles, which have demonstrated that they also contribute to fast-action release inherently. After 24 hours, half part releases 2.0 times as much mass of lidocaine per mg of particles and 0.9 times as much mass of dexamethasone per mg particles. We would expect to see that half part released the same amount of drug as weak API per particle during the sustained release period. We see this theory supported with release data from dexamethasone. However, this is not the case with lidocaine. This is likely due to the dissolution kinetics of lidocaine in the hydrogel lasting beyond 24 hours and having a stronger impact on the formula with less particles. This is reinforced by the relatively large crystal size of lidocaine that we observed. We expected that half part would release half as much overall drug during sustained release compared to weak API as it contains half the mass of particles. The experimental data shows this is the case in regard to dexamethasone. However, half part released nearly the same amount of lidocaine overall after 24 hours. Again, we attribute this to the dissolution kinetics of lidocaine lasting beyond 24 hours, in addition to half part containing less particles. Furthermore, this may be due to the effects of increasing the concentration of particles in the formulation. As noted by Wang et al. [33] particles affect the “pore” size of the hydrogel in regard to the “pore theory” which governs the diffusivity of PVA hydrogels formed by freeze-thaw cycle method. The presence of particles both limits the ability of large “pores” to form in the hydrogel and take up “pore” space themselves. With more particles exacerbating both of these effects further, we would expect to see less “free volume” in a hydrogel, which leads to reduced space for water to infiltrate the hydrogel and less diffusion as a result. These results support the theory that fast-action release can reliably be tuned by particle mass fraction independent of fast-action release.
We also wanted to see the absolute mass of drug released for weak API and half part. In the first 24 hours, weak API releases 1.3 times more mg lidocaine and 1.7 times more mg dexamethasone. After 24 hours, half part releases 1.1 times more mg lidocaine and 2.5 times less mg dexamethasone. In both fast-action and sustained release, weak API releases more mass of drug than half part. We expected to see this occur as there are twice as many particles in weak API as there are in half part. While results were not as distinct as anticipated, the release study showed that increasing the particle mass fraction did increase the overall mass of drug released will not sharply affecting the amount of drug released per mg of particles. The release rate from particles remains reliable no matter large their mass fraction.
In all formulations, release of 95% of the maximum value was achieved by 5 days, indicated a relatively short release period. While not tested in this study, many works in literature demonstrate the sustained release period of similar particles can be altered by modifying the PLGA molecular weight by Raman [34], lactide: Glycolide ratio by Wu [35], particle size by Acharya et al. [36] and polydispersity by Chen [37]. For instance, by fractionating PLGA particles from the same emulsion, Chen et al. showed that release kinetics from different size particles are vastly different and the size ratio of particles in an emulsion influences the overall release kinetics. In one study performed by Gu. PLGA particles embedded in PVA hydrogels were 34.26 μm ± 13.44 μm with a range of 2 μm to 75 μm and achieved release of dexamethasone up to 6 months into the release study. Thus, if our formulations were to require longer or shorter release period, particle size, particle size distribution or other parameters may be modified as required, while still allowing the fast-action release period to be altered through other means independently.
Our smaller particles likely contributed to a shorter release period. The drug release per mg of particles was not consistent in situations where it was expected (i.e. weak API vs. half part, during all sustained release periods). We assumed that particles were uniformly mixed into the hydrogel during the formation of composite molds. An analysis of our SEM imaging showed that the particles used in this study had. Once the composite molds went through the freeze-thaw cycle and were divided into 3 parts, it was not possible for us to measure the true mass of particles in each third. The same is true of the raw API mixed directly into the hydrogel. This may have led to high variance of drug release per mg of particles among the 3 pieces of the original formulation mold, especially with lidocaine, which had a visibly larger crystal size than dexamethasone.
The potential applications of a multi-drug dual-phase drug delivery system are numerous. One instance, which is of chief focus in relation to this work, is implantable devices. This type of composite with its anti-inflammatory payload could be coated onto an implantable device, such as an insulin infusion catheter, to combat the FBR caused by implantation [27,30]. This dual phase fast-action-then-sustained release profile has been shown to be effective for this purpose when used on glucose sensors [24]. Additionally, the use of two rates of drug release has uses in the field of cancer therapy, especially with the ability of this system to be delivered locally [10,23].
Conclusion
Our team created a drug delivery system capable of delivering two anti-inflammatory drugs, lidocaine and dexamethasone, at two different rates. The system consists of PLGA micro particles mixed into a PVA hydrogel matrix, creating a particle-hydrogel composite. The initial phase of release, known as fast-action release, is governed by the concentration of drug in the hydrogel. The following phase of release, known as sustained release, is governed by the size of particles. The absolute amount of drug delivered during sustained release can be altered using the fraction of particles relative to the entire system. Our results support these hypotheses. This study reveals the potential of creating multi-phase drug delivery systems that can be used to improve wound healing by combatting the foreign body response.
Acknowledgements
We would like to thank the Singh center for nanotechnology for their contribution to this study.
References
- Adam B (2009) Trastuzumab based therapy for patients with Her2-positive breast cancer. Am J Clin Oncol 33: 186-95. [Crossref], [Google Scholar], [Indexed]
- Guo HX, Shi YP (2009) A novel zein-based dry coating tablet design for zero-order release. Int J Pharm 370: 81–86. [Crossref], [Google Scholar], [Indexed]
- Ali M, Horikawa S, Venkatesh S, Saha J, Hong JW, et al. (2007) Zero-order therapeutic release from imprinted hydrogel contact lenses within in vitro physiological ocular tear flow. J Control Release 124: 154–162. [Crossref], [Google Scholar], [Indexed]
- Das NG, Das SK (2003) Controlled-release of oral dosage forms. Formul Fill Finish 15: 10–16. [Crossref], [Google Scholar]
- Sakurai M, Naruto I, Matsuyama K (2008) Evaluation for zero-order controlled release preparations of nifedipine tablet on dissolution test, together with cost benefit point of views. Yakugaku Zasshi 128: 819–826. [Crossref], [Google Scholar], [Indexed]
- Abrahams RA, Ronel SH (1975) Biocompatible implants for the sustained zero-order release of narcotic antagonists. J Biomed Mater Res 9: 355–366. [Crossref], [Google Scholar]
- Varelas CG, Dixon DG, Steiner CA (1995) Zero-order release from biphasic polymer hydrogels. J Control Release 34: 185-192. [Crossref], [Google Scholar]
- Yang L, Fassihi R (1996) Zero-order release kinetics from a self-correcting floatable asymmetric configuration drug delivery system. J Pharm Sci 85: 170-173. [Crossref], [Google Scholar], [Indexed]
- Donbrow M, Samuelov Y (1980) Zero order drug delivery from doublea? layered porous films: release rate profiles from ethyl cellulose, hydroxypropyl cellulose and polyethylene glycol mixtures. J Pharm Pharmacol 32: 463–470. [Crossref], [Google Scholar], [Indexed]
- Ranganath SH, Kee I, Krantz WB, Chow PKH, Wang CH (2009) Hydrogel matrix entrapping PLGA-paclitaxel microspheres: Drug delivery with near zero-order release and implantability advantages for malignant brain tumour chemotherapy. Pharm Res 26: 2101-2114. [Crossref], [Google Scholar], [Indexed]
- Berkland C, King M, Cox A, Kim K, Pack DW (2002) Precise control of PLG microsphere size provides enhanced control of drug release rate. J Control Release 82: 137–147. [Crossref], [Google Scholar], [Indexed]
- Huang X, Brazel CS (2001) On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release 73: 121-136. [Crossref], [Google Scholar], [Indexed]
- Yamaguchi Y, Takenaga M, Kitagawa A, Ogawa Y, Mizushima Y, Igarashi R (2022) Insulin-loaded biodegradable PLGA microcapsules: Initial burst release controlled by hydrophilic additives. J Control Release 81: 235-249. [Crossref], [Google Scholar], [Indexed]
- Mallapragada SK, Peppas NA, Colombo P (1997) Crystal dissolution-controlled release systems. II. Metronidazole release from semi crystalline poly (vinyl alcohol) systems. J Biomed Mater Res 36: 125-130. [Crossref], [Google Scholar], [Indexed]
- Hasan AS, Socha M, Lamprecht A, El Ghazouani F , Sapin A, etal. (2007) Effect of the microencapsulation of nanoparticles on the reduction of burst release. Int J Pharm 344: 53-61. [Crossref], [Google Scholar], [Indexed]
- Hezaveh H, Muhamad II (2013) Controlled drug release via minimization of burst release in pH-response kappa-carrageenan/polyvinyl alcohol hydrogels. Chem Eng Res Des 91: 508-519.[Crossref], [Google Scholar]
- Huang YY, Chung TW, Tzeng TW (1999) A method using biodegradable polylactides/polyethylene glycol for drug release with reduced initial burst. Int J Pharm 182: 93-100.[Crossref], [Google Scholar], [Indexed]
- Narasimhan BRL (1997) Abstracts and undefined on the importance of the burst effect during drug release from polymer films Chem SOC 1155. [Crossref], [Google Scholar], [Indexed]
- Chien YW (1988) Controlled release of biologically active agents. By Richard W. Baker.
- Patil NS, Dordick JS, Rethwisch DG (1996) Macroporous poly(sucrose acrylate) hydrogel for controlled release of macromolecules. Biomaterials 17: 2343–2350. [Crossref], [Google Scholar], [Indexed]
- Lischer S (2011) Antibacterial burst-release from minimal Ag-containing plasma polymer coatings. J R Soc Interface 8: 1019–1030. [Crossref], [Google Scholar], [Indexed]
- Setterstrom JA, Ticet TR, Myerst WE, Development of encapsulated antibiotics for topical administration to wounds. [Crossref], [Google Scholar]
- Xu S, Wang W, Li X, Liu J, Dong A, Deng L (2014)Sustained release of PTX-incorporated nanoparticles synergized by burst release of DOXÁHCl from thermosensitive modified PEG/PCL hydrogel to improve anti-tumor efficiency. Eur J Pharm Sci 62: 267-273. [Crossref], [Google Scholar], [Indexed]
- Kastellorizios M, Papadimitrakopoulos F, Burgess DJ (2015) Prevention of foreign body reaction in a pre-clinical large animal model. J Control Release 202: 101–107. [Crossref], [Google Scholar], [Indexed]
- Rawat A, Burgess DJ (2011) Effect of physical ageing on the performance of dexamethasone loaded PLGA microspheres. Int J Pharm 415: 164–168. [Crossref], [Google Scholar], [Indexed]
- Zolnik BS, Raton JL, Burgess DJ (2005) Application of USP apparatus 4 and in situ fiber optic analysis to microsphere release testing. Dissolution Technol 12: 11-14. [Crossref], [Google Scholar]
- Wang Y, Papadimitrakopoulos F, Burgess DJ (2013) Polymeric 'smart' coatings to prevent foreign body response to implantable biosensors. Control. Release 169: 341–347. [Crossref], [Google Scholar], [Indexed]
- Chen PC, Kohane DS, Park YJ, Bartlett RH, Langer R, Yang VC (2004) Injectable microparticle-gel system for prolonged and localized lidocaine release. II. In vivo anesthetic effects. J Biomed Mate Res A 70: 459–66. [Crossref], [Google Scholar], [Indexed]
- Bhardwaj U, Sura R, Papadimitrakopoulos F, Burgess DJ (2010) PLGA/PVA hydrogel composites for long-term inflammation control following s.c. implantation. Int J Pharm 384: 78–86. [Crossref], [Google Scholar], [Indexed]
- Kwangjae C, Xu W, Shuming N, Georgia CZ, Shin(2008) Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res 14: 1310-1316. [Crossref], [Google Scholar], [Indexed]
- Lai F, Pini E, Angioni G et al. (2011) Nanocrystals as tool to improve piroxicam dissolution rate in novel orally disintegrating tablets. Eur J Pharm Biopharm 79: 552-558. [Crossref], [Google Scholar], [Indexed]
- Robert SC, Deborah C, Barbara K, Ramin K, Mohammad KF (2008) Aiming for the heart: Targeted delivery of drugs to diseased cardiac tissue. Expert Opin Drug Deliv. 5: 459-70. [Crossref], [Google Scholar], [Indexed]
- Raman C, Berkland C, Kim K, Pack DW (2005) Modeling small-molecule release from PLG microspheres: Effects of polymer degradation and nonuniform drug distribution. J Control Release 103: 149-158. [Crossref], [Google Scholar], [Indexed]
- Wu XS, Wang N (2001) Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers. Part II: Biodegradation. J Biomater Sci Polym Ed 12: 21-34. [Crossref], [Google Scholar], [Indexed]
- Acharya G, Shin CS, Vedantham K et al. (2010) A study of drug release from homogeneous PLGA microstructures. J Control Release 146: 201-206. [Crossref], [Google Scholar], [Indexed]
- Chen W, Palazzo A, Hennink WE, Kok RJ (2017) Effect of particle size on drug loading and release kinetics of gefitinib-loaded PLGA microspheres. Mol Pharm 14: 459-467. [Crossref], [Google Scholar]
- Gu B, Papadimitrakopoulos F, Burgess DJ (2018) PLGA microsphere/PVA hydrogel coatings suppress the foreign body reaction for 6 months. J Control Release 289: 35-43. [Crossref], [Google Scholar]