Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 2 Issue: 1
Population Pharmacokinetics of Lixisenatide, a Once-Daily Human Glucagon-Like Peptide-1 Receptor Agonist, in Healthy Subjects and in Patients with Type 2 Diabetes
| Thomas Frank* |
| Sanofi-Aventis Deutschland GmbH, Frankfurt am Main, Germany |
| Corresponding author : Frank T Sanofi-Aventis Deutschland GmbH, Clinical Pharmacokinetics/Pharmacometrics, Drug Disposition, Safety and Animal Research, Industriepark Höchst, D-65926 Frankfurt am Main, Germany Tel: +49 69 305 30230; Fax: +49 69 305 331723 E-mail: thomas.frank@sanofi.com |
| Received: December 19, 2012 Accepted: January 28, 2013 Published: January 30, 2013 |
| Citation: Frank T (2013) Population Pharmacokinetics of Lixisenatide, a Once-Daily Human Glucagon-Like Peptide-1 Receptor Agonist, in Healthy Subjects and in Patients with Type 2 Diabetes. J Pharm Drug Deliv Res 2:1. doi:10.4172/2325-9604.1000112 |
Abstract
Population Pharmacokinetics of Lixisenatide, a Once- Daily Human Glucagon-Like Peptide-1 Receptor Agonist, in Healthy Subjects and in Patients with Type 2 Diabetes
Lixisenatide is a potent and selective glucagon-like peptide-1(GLP-1) receptor agonist developed for the treatment of type 2 diabetes (T2DM) with a subcutaneous once-daily regimen. The substance is currently under review for marketing authorization for the management of T2DM. In this study, the population pharmacokinetics in healthy volunteers and in adult T2DM patients was investigated.
Keywords |
|
| Lixisenatide; Population pharmacokinetics; Type 2 diabetes; Glucagon-like peptide-1 | |
Abbreviations |
|
| SD: Standard Deviation; CWRES: Weighted Residuals Evaluated at Individual Conditional Estimates; DOBJ: Difference of the Objective Function Values Between Reduced and Full Model or Likelihood-Ratio-Test; ka: Absorption Rate Constant; ke: Elimination Rate Constant; AUC: Area Under the Curve; BID: Twice Daily; BMI: Body Mass Index; CLCR: Creatinine Clearance; CI: Confidence Interval; CL/F: Apparent Clearance; Cmax: Maximum Concentration; CV: Coefficient of Variation; F: Bioavailability; IIV: Inter-Individual Variability; IOV: Inter-Occasion Variability; LLOQ: Lower Limit of Quantification; MAT: Mean Absorption Time; PNWT: Predicted Normal Weight; QD: Once Daily; SE: Standard Error; t1/2z: Apparent Terminal Half-Life; tmax: First Time to Reach Maximum Concentration; V/F: Apparent Volume Of Distribution; WT: Body Weight; η: Random Inter-Individual Variability - Unexplained Difference Between an Individual, Fitted Parameter Value and the Fitted Parameter Value of the Structural Model (Population Mean Value); θ: Vector of PK Parameter Describing the Fixed Effect Model. | |
Introduction |
|
| The glucagon-like peptide-1 (GLP-1) receptor is widely expressed in the pancreas and the gastrointestinal tract and represents an established therapeutic target in type 2 diabetes mellitus (T2DM). The incretin hormone GLP-1, a physiological GLP-1 receptor agonist, is secreted by intestinal endocrine L-cells in response to food and enhances meal-stimulated insulin secretion — the so-called “incretin effect” [1,2]. As a class, GLP-1 and its analogues have a beneficial impact on the metabolism of nutrients and are known to stimulate insulin release from the pancreatic islets (insulinotropic release) in a glucose-dependent manner, suppress glucagon secretion, increase insulin sensitivity, delay gastric emptying, and reduce appetite feelings [3]. As a result of these characteristics, these compounds are effective in lowering fasting and postprandial blood glucose, and provide a long-term benefit for glycemic control associated with a low risk of hypoglycemia and the potential for weight reduction. The use of GLP-1 receptor agonists is endorsed as an add-on treatment for T2DM [4,5]. | |
| Lixisenatide is a synthetic GLP-1 receptor agonist structurally based on exenatide with a modified C-terminus of six lysine residues, which means it is able to withstand physiological degradation by dipeptidyl peptidase-4 (DPP-4) and, therefore, prolongs the physiological effects of GLP-1 itself [6]. The affinity of lixisenatide for the human GLP-1 receptor is approximately four times higher compared to native GLP-1 [7]. | |
| Lixisenatide, developed as subcutaneous once-daily regimen, is currently under review for marketing authorization for the management of T2DM. The efficacy and safety of once-daily lixisenatide has been assessed in the GetGoal phase 3 clinical trial program. Results have shown beneficial effects on HbA1c compared with placebo as add-on to commonly used antidiabetes agents, with limited risk of hypoglycemia and with beneficial effect on body weight [8]. | |
| So far, data on the population pharmacokinetics (PPK) of lixisenatide has not been fully published. In particular, there is no single model describing the PK and covariate effects in the target population as well as in healthy volunteers. PPK analysis is a robust tool for obtaining valuable pharmacokinetic (PK) information from large clinical trials under conditions of sparse concentration sampling; its importance lies in the testing of the influence of potential covariates on the pharmacokinetics by incorporating patient-specific information into the modeling process [9]. Data from the current GetGoal phase 3 clinical trial program [8,10] provided the opportunity to determine PPK parameters for lixisenatide in a T2DM patient population and evaluate the effect of potential covariates (e.g., age, body weight, sex, and race) on lixisenatide exposure. | |
| The purpose of the present analysis is to characterize the PPK of lixisenatide in healthy volunteers and in T2DM patients, and to identify subject characteristics that are predictive of variability in the pharmacokinetic parameters of lixisenatide. | |
Methods |
|
| Study design and subjects | |
| The data included in the pooled analysis originates from 7 clinical studies: two phase 1 and two phase 2 studies as well as three phase 3 studies. The relevant characteristics of each clinical study included in the current analyses are summarized in Table 1. In these studies, lixisenatide was administered as a single subcutaneous (s.c.) dose (n=75) or multiple s.c. doses (n=1148), generally 30 minutes before meals (breakfast for once daily [QD] administration, breakfast and dinner for twice daily [BID] administration) unless specified otherwise in Table 1. | |
| Table 1: Summary of study data and subject demographics used in the population pharmacokinetic analysis. | |
| The studies were conducted in accordance with good clinical practice guidelines and the Declaration of Helsinki and its revisions. The trial protocols and informed consent documents were all reviewed and approved by independent ethics committees. Written informed consent was provided by each subject or their legal guardian. | |
| Pharmacokinetic assessments | |
| Total lixisenatide, i.e., bound and unbound to anti-lixisenatideantibodies, in human ethylenediaminetetraacetic acid (EDTA) plasma was measured by a validated double-antibody sandwich enzymelinked immunosorbent assay (ELISA) technique with a lower limit of quantification (LLOQ) of 12 ng/l. For study POP6053, the same assay was used, but the LLOQ value had to be corrected retrospectively to 8.8 ng/l. Since this is only a mathematical correction, the validated precision and accuracy of the assay is not influenced. At the LLOQ, the coefficients of variation of accuracy and precision were <10% (in study POP6053 ≤ 25%). Anti-lixisenatide antibodies in studies BDR6864, POP6053, PDY6797 and DRI6012 were measured with a radioimmunoprecipitation method. Anti-lixisenatide antibodies in studies EFC6018, EFC10887 and EFC6015 were measured with a validated surface plasmon resonance (SPR) method on Biacore. Patients were classified as having a positive anti-lixisenatide antibody status if antibodies to lixisenatide were detected. | |
| Lixisenatide PK is greatly influenced by presence of lixisenatide antibodies. In both healthy volunteers and patients lixisenatide AUC and Cmax are markedly increased, tmax delayed and half-life prolonged in anti-lixisenatide antibody positive patients. This is accompanied by a large increase in inter-individual variability. The interference of antibody presence with the PK of lixisenatide is difficult to handle in modeling as the individual predictions become impossible. Therefore, all visits associated with a positive or missing anti-lixisenatide antibody status were discarded in the analysis. | |
| Samples below the LLOQ (BLQ) were either discarded (in studies EFC6018, EFC6015 and EFC10887, method “M1” in [11]) or treated according to method “M6” in phase 1 and 2 studies, i.e,. each BLQ observation x after Cmax was replaced by LLOQ/2, except that any and all consecutive BLQ observations succeeding x were discarded (as with method “M1”). | |
| Population pharmacokinetic analysis | |
| NONMEM, version 7.2 (ICON Development Solutions, Ellicott City, MD), was used to conduct all PPK analyses. NONMEM, and its modules NM-TRAN and PREDPP, were compiled with the GNU Compiler Collection for Fortran 90/95 (GCC 4.5) [12] running under openSUSE Linux, version 11.4, on a computer cluster [13]. The firstorder conditional estimation method with η – ε interaction (FOCE-I) was applied throughout after logarithmic data transformation. The R software [14] was used to process the data and to perform graphic and statistical analysis. An in-house developed R library was used to facilitate NONMEM processing. Additional software tools used were Xpose [15] and Perl-speaks-NONMEM [16]. | |
| Structural and pharmacostatistical model: A one-compartment model with first-order absorption (PREDPP subroutines ADVAN2/TRANS1) was used to describe the lixisenatide plasma concentration–time profiles. The structural model was parameterized using the mean absorption time (MAT), which is approximately 1/ka, the first-order absorption rate constant, apparent volume of distribution (V/F), and apparent clearance (CL/F). The bioavailability, F, was unknown. | |
| The random effects of PPK included IIV (inter-individual variability), IOV and residual error. The IIV and IOV were investigated for all pharmacokinetic parameters including F. The distribution of the parameters was assumed log-normal, so that exponential models were used to account for IIV and IOV: | |
| ln(Pi,j)= ln(PTv)+ηi+ηi,j (1) | |
| where Pi,j is the estimated parameter value for a given individual i at the jth occasion, PTV is the typical population value of the parameter, ηi describes the variation of individual i from the population estimate (that is, IIV), and ηi,j represents the variability of occasion j (that is, IOV). In all cases, η is assumed to be normally distributed: ηi~N(0,ω2) and ηi,j~N(0,ωIOV2), with inter-individual variance-covariance matrix Ω. An “occasion” was defined as a group of sequential dosing records within one individual terminated by at least one observation event (concentration), and the variances were considered to be sampled from the same distribution. The IIV and the IOV, respectively, are approximately equal to a CV (coefficient of variation [%]). | |
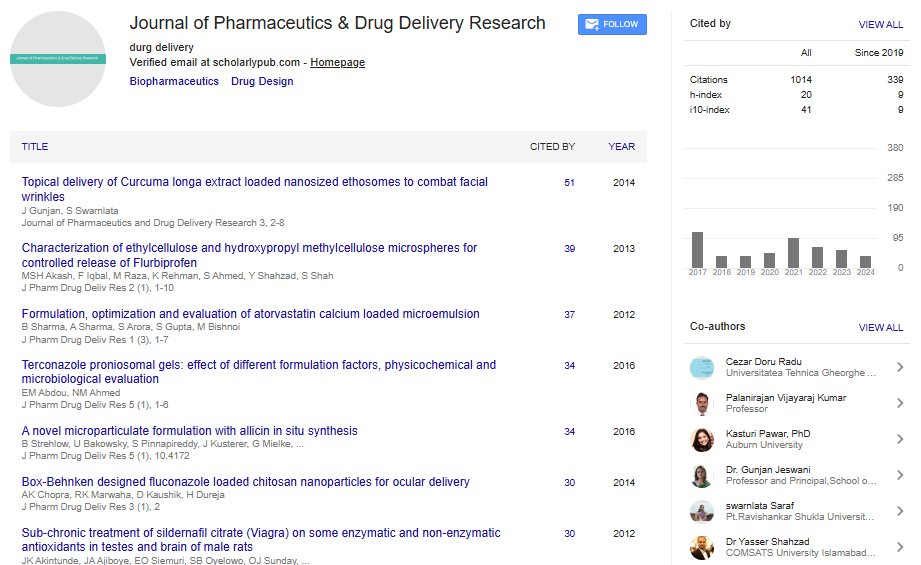
| The residual unexplained variability representing the variance between the observed plasma concentrations (Y) and those predicted by the model (IPRED) were estimated as follows: | |
| ln(Y) = ln(IPRED + M) + W·ε (2) | |
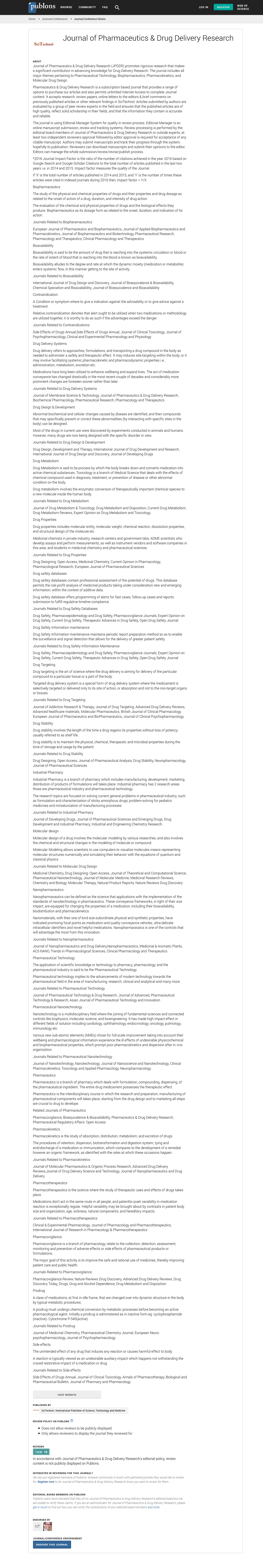
| with ε~N(0,1) and with the intra-individual error model in equation 3. | |
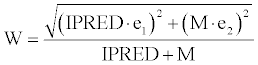 (3) (3) |
|
| The additive constant M, the quasi-proportional error e1, and the quasi-additive error e2 were fitted parameters. Reduction of the residual error model to a simpler form was not tested during the model development process. | |
| The model judged to be the best structural model on the basis of the goodness-of-fit plots, 95% confidence interval of the parameter estimate, agreement with the non-compartmentally calculated pharmacokinetic parameters, and the likelihood ratio test. | |
| Covariate evaluation: Before the covariate search in NONMEM scatter and box-and-whisker plots of the individual η versus covariates helped to identify potential correlations. | |
| Covariates with potential impact on the parameters of the PPK model, that is age, body weight (WT), body mass index (BMI), predicted normal weight (PNWT), serum creatinine concentration (CREA), CLCR, SGOT (serum glutamate oxalo-acetate transaminase), SGPT (serum glutamate pyruvate transaminase), dose, regimen (i.e., QD or BID), gender, and race were identified directly in NONMEM in a stepwise manner, initially evaluated by univariate analysis followed by a comprehensive forward addition backward elimination procedure to build the final covariate model. In the univariate analysis, a rank was established to decide the order each covariate would be incorporated into the model, based on the change of the objective function (and the associated P value) with respect to the base model. The covariates CLCR, SGOT, SGPT, and CREA were only tested on CL/F. As far as possible, summary variables were calculated to represent correlated predictors, e.g., BMI reflects both weight and body height; PNWT, a parameter especially developed for use in obese subjects [17] is a correction of lean body weight (LBW). PNWT is approximately the body weight for patients with BMI <30 kg/m2 but is significantly lower for obese patients with BMI >30 kg/m2. CLCR was calculated according to the Cockcroft-Gault equation [18]. The covariate CLCRN where WT is substituted by PNWT in the Cockroft-Gault equation was also tested on CL/F. SGOT and SGPT were considered as rough markers of hepatic function. In general, when several covariates are markers of the same feature and/or they correlated with each other, the best one was chosen based upon likelihood-ratio test. | |
| Continuous covariates were included in the model using equations 4 and 5. | |
| PTV=θx.(1+θy.(Covariate- Covariate median)) (5) | |
| Other relationships, e.g., exponential, were also tried. The discrete covariates (that is, race, treatment regimen and gender) were introduced using if/else statements. | |
| After the forward step, each covariate in the full model was tested in turn by removing each entity one by one to confirm the statistical significance. The model with only significant covariates was chosen as the pre-final model (backward elimination step). To be retained in the final model the 95% confidence interval on the covariate effect (based on the standard error of the parameter estimate given by NONMEM) should not include the null parameter. Outliers, identified as those observations with absolute conditional weighted residual values (CWRES) greater than 6, were excluded from the data set, and the model was reassessed. Once this process was completed, the resultant model was considered as the final PPK model. | |
| Model evaluation: The PPK model was evaluated on the basis of goodness-of-fit plots, 95% confidence interval for the parameter estimates, and the likelihood ratio test. The 95% confidence interval (CI) for parameter estimates was obtained from the point estimate ± 2 × standard error (SE), which was taken from the covariance step. The coefficients of variance (%CV) for IIV, IOV, and intra-individual variability were calculated from the square root of the variance. In the likelihood ratio test, the objective function value difference (ΔOBJ) was used for evaluating the statistical significance of the parameters. The p values for the forward selection step and backward elimination step were <0.01 (ΔOBJ was >6.63) and P<0.001 (ΔOBJ was >10.83), respectively. | |
| A prediction- and variability-corrected visual predictive check (VPC) was performed as a model validation technique [19]. One thousand new datasets with the same subjects,, dosing history, number of observations, sampling scheme, and covariate values as in the pooled analysis were simulated with the estimated final model parameters. Descriptive statistics of simulated plasma lixisenatide concentrations were then compared with observed plasma lixisenatide concentrations. | |
| For the final model, the individual predicted PK parameters (Pi) were computed from the population parameters (PTV) using all available sources of information (i.e., with individual covariates if any) to judge the predictive performance. The bias (mean predicted error, ME) and precision (root mean squared error, RMSE), and their associated 95% CIs on the PK parameters CL/F, V/F and MAT were evaluated according to the following equation [20]: | |
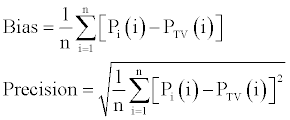 (6) (6) |
|
| In these expressions the index i refers to the individual parameter,
and n is the total number of values. Bias and precision were also
expressed as a percentage of the mean Pi value. Optimally the bias and precision are not different from zero. |
|
Results and Discussion |
|
| The initial data set consisted of 9623 total lixisenatide plasma concentrations from 1223 individuals (75 healthy subjects and 1148 patients with T2DM). The number of plasma concentrations that were either classified as antibody-positive or as BLQ (after processing with the BLQ substitution rule as described above) was 3853, so that 5770 concentration values (‘observations’) remained for modeling. | |
| For antibody-positive samples, the total lixisenatide (bound and unbound to anti-lixisenatide-antibodies) was determined using pre-assay sample processing for the dissociation of the lixisenatideanti- lixisenatide-antibody immune-complexes. That means, that the lixisenatide concentration data were insufficient to distinguish between bound and unbound lixisenatide; therefore, it was not possible to develop a PPK model that included these two components, and a substantial portion of observations was disregarded in the analysis as they were associated with the presence of anti-lixisenatide antibodies. As a peptide, lixisenatide may be associated with antibody formation after repeated dosing: The proportion of anti-lixisenatide antibody-positive subjects increased to about 70% after 24 weeks of treatment, even though most of them had a low level. In the phase 2 and 3 studies, the concentrations of total lixisenatide (lixisenatide unbound and bound to antibodies) were increased in the presence of anti-lixisenatide antibodies, with accompanying increases in inter-individual variability. To model, not only the total lixisenatide concentrations and the determination of antibody concentrations at a few occasions would have been present, but also information about affinities and their development over time. | |
| To search for potential outliers in the initial dataset, a onecompartment model with IIV on CL/F, V/F and MAT was initially fitted to the data. It was found that there were 162 observations that were higher than twice the LLOQ and for which the log-transformed concentrations exceeded the IPRED by more than 2. These outlying observations were almost entirely pre-dose values and were seen as strong evidence that the corresponding samples were anti-lixisenatide antibody-positive, even if this had not been determined analytically. The identified outliers were excluded in all subsequent fits. No subject had to be completely excluded as a result of the removal of outlier data. | |
| The resulting dataset of the above described operation was taken as the total dataset, and it consisted of 5608 observations (antibody-negative samples above the LLOQ plus those BLQ samples transformed to LLOQ/2, as described under ‘Pharmacokinetic assessments’). The number of samples below the LLOQ in the total data set was 2738 (32.8%). | |
| A summary of selected demographic characteristics of the total dataset, i.e., the PPK population, can be found in Table 1. The mean age of subjects studied was 56.5 years, and 46.9% were females. The number of Caucasian, Black, Asian/Oriental and other ethnic origin in the pooled analysis was 729 (59.6%), 40 (3.3%), 398 (32.5%) and 56 (4.6%), respectively. There were 16 subjects (1.3%) who were older than 75 years, and there were 22 subjects (1.8%) who weighed 50 kg or less. In contrary, 652 subjects (53.5%) weighed more than 80 kg, and 48.5% of the participants were obese (BMI>30 kg/m2). | |
| The IOV was the main model component that reduced the objective function values most in the search for the base model. Attempts to model IIV or IOV additionally on F, also in combination with IOV on V/F, CL/F, and MAT, were not successful. | |
| Significant covariates that explained IIV in CL/F identified in the univariate analysis using NONMEM (P<0.01) included CLCR, SGPT, and dose. Covariates on CL/F that are markers of the same feature, but led to a smaller reduction of OFV (e.g., CLCRN, BMI) were not further considered. For V/F, covariates that explained IIV included WT, race and age. Additionally, the univariate analysis identified BMI, dose and race as explaining IIV in MAT. All significant covariates were then included in the base model in a forward stepwise manner until there was no further reduction in the objective function value. The full model included WT on V/F, CLCR and SGPT on CL/F, and dose and BMI on MAT. | |
| With the full covariate model, the effect of each explanatory covariate was re-assessed through a stepwise backward deletion approach. As a result of the model refinement, SGPT was removed from the model as covariate on CL/F. All of the covariates that remained in the model (WT on V/F, CLCR on CL/F, and dose and BMI on MAT) significantly contributed to the model fitting. The influence of BMI on MAT modeled as fractional change of population MAT per kg/m2 deviation from the median BMI, however, was determined inaccurately with 0.009 (95% Cl: -0.001 to 0.018) and was removed from the model. The IOV of MAT was also removed from the model due to a large %CV of its variance (56%). | |
| After incorporating the covariates into the base model, the IIV on CL/F increased, and the IIV for MAT only decreased from 33.2% to 30.0%, whereas the IIV for V/F clearly decreased from 61.4% to 30.3% (all values in log domain, Table 2). Thus there were essentially no relevant reductions in the IIV for CL/F and MAT by incorporating covariates. This is not surprising because the intrinsic and extrinsic factors that determine the disposition of lixisenatide are not fully understood. Random effects for lixisenatide clearance (ηCL) by continuous or categorical variables for the final model (Figure 3) showed no systematic trend or bias. | |
| Table 2: Parameter estimates from the final population pharmacokinetic model. | |
| Lixisenatide CL/F was shown to be affected by renal function (CLCR). The model predicted that an individual weighing 82.1 kg would have CL/F decreased by 9.9% when renal clearance is reduced by 1 l/h, as compared with the population median. An individual weighing 82.1 kg with moderate renal impairment (CLCR of 1.8 to 3.0 l/h) may therefore have a 1.5- to 1.8-fold increase in exposure if lixisenatide is administered at a dose of 20 μg. The inclusion of CLCR in the model for lixisenatide CL/F has some mechanistic validity because principal elimination of lixisenatide takes place via proteolytic tubular breakdown in the kidneys. Renal function declines with advancing age because of physiological reductions in glomerular filtration rate and renal tubular secretion. Age, body weight, and gender are already part of the Cockroft-Gault formula, so that, after inclusion of CLCR, these were not kept in the model as significant, isolated covariates. | |
| In seven individuals outliers were identified based on CWRES in the penultimate fit. These were apparently faulting observations somewhere in their lixisenatide concentration-time profiles. No subjects were entirely excluded as a result of the removal of data with high-weighted residuals. From the final input data set without these outliers, the estimates of the fixed effect and random effect parameters were almost the same as those for the data set with outliers. The run without the described 7 outliers, in the text referred to as the final model, is summarized in Table 2. It can be seen that the MAT of 3.25 hours was longer than the population mean elimination time (V/CL) of 1.07 hours. The absorption limitation was still the case for the large majority of participants when comparing individual parameters. In this flip-flop kinetic situation, the t1/2z for most individuals corresponded to the absorption process and had to be calculated as ln 2 × MAT ~ 2.3 hours (“flop” situation). In six subjects only, i.e., in 0.5% of the cases, ka was greater than ke (“flip” situation). | |
| The corresponding goodness-of-fit plots are shown in Figure 1. Individual plots of observed, individual predicted and population predicted concentrations versus time after dose that illustrate how well the final model describes the data for selected individuals are presented in Figure 2. Absorption kinetics of peptides like lixisenatide (molecular mass of 4858.6 g/mole) from subcutaneous administration has been shown to be highly dependent on the site of injection, temperature, and degree of rubbing at the injection site [18]. Figure 2 shows that this finding could be reproduced in the present analysis. The large IOV (45% for CL/F, 59% for V/F) suggested a limited potential for dose-optimization of this regimen. Because targeted clinical effects on HbA1c need weeks of lixisenatide treatment, the effect of any daily fluctuation of the drug input caused by the IOV is balanced out in the long-term and therefore, probably, without clinical relevance. | |
| Figure 1: Final model: goodness-of-fit plots for lixisenatide. | |
| Figure 2: Plot of population and individual predicted lixisenatide concentrations in the final model versus the measured lixisenatide concentrations for selected participants. A: subject no. 38 injecting lixisenatide at three different sites of absorption (study BDR6864). B: subject nos. 1204, 1208, 1212 and 1217 with different degrees of renal impairment in study POP6053. C: subject no. 774 of study EFC6015 at four different occasions. | |
| Figure 3: Random effects for lixisenatide clearance (ηCL) by continuous or categorical variables for the final model. The blue line represents the smooth of the data. | |
| The results show a significant mean bias for CL/F, and V/F, but not for MAT (Table 3). The bias remained however below 20% of the mean. The VPC in Figure 4 illustrates, that with several exceptions, most of the observed concentrations fell within the 5th to 95th percentiles of simulated values indicating a reasonably well description of the observed lixisenatide concentrations. | |
| Figure 4: Prediction and variance-corrected visual predictive check for the final model. The pink areas indicate the simulation-based 90% confidence interval for the model-predicted 5%, 50%, and 95% percentiles. The violet lines indicate the observed 5%, 50% and 95% percentiles. The red lines connect the median of the model-predicted percentiles. The blue circles are the prediction-corrected observations. | |
| Table 3: Bias, precision and 95% CIs of individual predicted (Pind) vs. population parameters (Ppop) in the final model. | |
Acknowledgements |
|
| I acknowledge the contribution of all Sanofi staff, investigators, and nursing staff to the planning and execution of the trials used in this paper. I would like to thank especially my colleagues Dr D. Rueppel and Dr M. Burschka for their support in data management. | |
Declaration of Interest |
|
| The author is employee of Sanofi-Aventis Deutschland GmbH, Germany. | |
References |
|
|
|