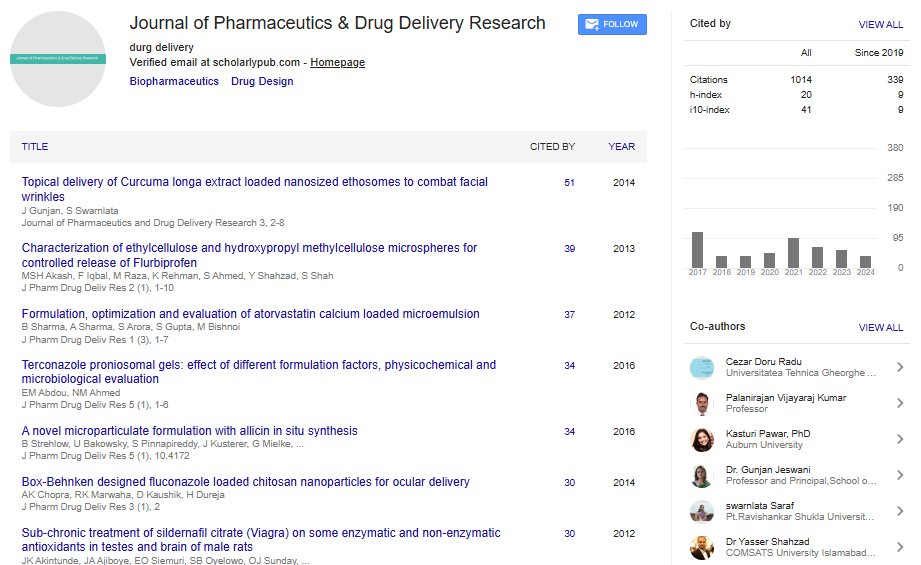
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Drug Deliv Res Vol: 4 Issue: 1
Fludarabine- (C2-methylhydroxyphosphoramide)- [anti-IGF-1R]: Synthesis and Selectively Targeted Anti-Neoplastic Cytotoxicity against Pulmonary Adenocarcinoma (A549)
| Coyne CP1,2* and Lakshmi Narayanan1 |
| 1Department of Basic Sciences, College of Veterinary Medicine, Wise Center, Mississippi State University, Mississippi State, Mississippi, USA |
| 2College of Veterinary Medicine, Mississippi State University, Mississippi, USA |
| Corresponding author : CP Coyne College of Veterinary Medicine, Wise Center, Mississippi State University, Mississippi State, Mississippi 39762, USA Tel: +662 325-1120 E-mail: coyne@cvm.msstate.edu |
| Received: November 25, 2014 Accepted: March 16, 2015 Published: June 20, 2015 |
| Citation: Coyne CP, Narayanan L (2015) Fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]: Synthesis and Selectively “Targeted†Anti-Neoplastic Cytotoxicity against Pulmonary Adenocarcinoma (A549). J Pharm Drug Deliv Res 4:1. doi:10.4172/2325-9604.1000129 |
Abstract
Fludarabine- (C2-methylhydroxyphosphoramide)-[anti-IGF-1R]: Synthesis and Selectively “Targeted”Anti-Neoplastic Cytotoxicity against Pulmonary Adenocarcinoma (A549)
Introduction: Many if not most conventional small molecular weight chemotherapeutics are highly potent against many forms of neoplastic disease. Unfortunately, majority of an administered dose unintentionally diffuses passively into normal tissues and healthy organ systems following intravenous administration. One strategy for both increasing potency and reducing dose-limited sequela is the selective “targeted” delivery of conventional chemotherapeutic agents.
Materials and Methods: The fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was synthesized by initially reacting fludarabine with a carbodiimide to form a fludarabine carbodiimide phosphate ester intermediate that was subsequently reacted with imidazole to create an amine-reactive fludarabine- (C2-phosphorylimidazolide) intermediate. Monoclonal anti-IGF-1R immunoglobulin was combined with the amine-reactive fludarabine- (C2-phosphorylimidazolide) intermediate resulting in the synthesis of covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic. Residual fludarabine and un-reacted reagents were removed by serial microfiltration (MWCO 10,000) and monitored by analytical-scale HP-TLC. Retained IGF-1R binding-avidity of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was established by cell-ELISA using pulmonary adenocarcinoma cell (A549) which over-expresses IGF-1R and EGFR. Anti-neoplastic cytotoxic potency of fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] was determined against pulmonary adenocarcinoma (A549) using an MTT-based vitality stain methodology.
Results: The fludarabine molar-incorporation-index for fludarabine- (C2-methylhydroxyphosphoramide)-[anti-IGF-R1] was 3.67:1 while non-covalently bound fludarabine was not detected by analytical scale HP-TLC following serial micro-filtration. Sizeseparation fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] by SDS-PAGE with chemo luminescent autoradiography detected only a single 150-kDa band. Cell-ELISA of fludarabine- (C2-methylhydroxyphosphoramide)-[anti-IGF-1R] measuring total immunoglobulin bound to exterior surface membranes of pulmonary adenocarcinoma (A549) increased with elevations
Keywords: Fludarabine,Immunoglobulin, Pulmonary adenocarcinoma
Keywords |
|
| Anti-neoplastic cytotoxicity; Pulmonary adenocarcinoma; Cytotoxic potency | |
Introduction |
|
| Fludarabine([(2R,3R,4S,5R)-5-(6-amino-2-fluoro-purin-9-yl)- 3,4-dihydroxy-oxolan-2-yl]methylhydroxyphosphonic acid) is a phosphorylated adenosine deaminase-resistant purine nucleotide analog that enters the cytosol of neoplastic cells by active transport as does methotrexate (5-FU enters by facilitated transport processes). The mechanisms-of-action for fludarabine are dependent upon it functioning as a biochemical substrate for deoxycytidine kinase which converts the chemotherapeutic to the primary active metabolite, 5’-fludarabine-ATP (F-ara-ATP) [1]. Anti-neoplastic cytotoxicity is primarily associated with competitive substitution for deoxyadenosine triphosphate (dATP) which profoundly inhibits DNA polymerase and terminates progressive DNA strand synthesis at the site of incorporation. In addition, the active metabolite F-ara- ATP suppresses ribonucleotide reductase biochemical activity thereby causing declines in cytosol dATP and further increases fludarabine potency. Neoplastic sub-populations residing within either rapidly dividing (M-phase) or “resting” (G0 phase) stages of the cell cycle are inhibited by fludarabine resulting in detectable concentrationdependent declines in viability and proliferation. Fludarabine is one of the most potent anti-metabolite class chemotherapeutics but the cytotoxic effect it evokes is not immediate which in part is a function of its known mechanism-of-action. Fludarabine anti-neoplastic cytotoxicity occurs as a result of continual inhibition of DNA/ RNA polymerase biochemical activity and its cumulative physical incorporation into more and more nucleotide strands leading to an eventual collapse of the neoplastic cell proteome. | |
| The plasma T1/2 half-life for fludarabine is approximately 10-hours and under normal physiological conditions it becomes ionically trapped within the intravascular compartment which affords some degree of specificity through body fluid compartment partitioning. The differential transport and level of phosphorylation for F-ara-A in concert with F-ara-ATP accumulation within normal and cancer cells may constitute the biochemical basis for its relatively greater margin-of-safety. The T1/2 plasma half-life for fludarabine is predominately determined by its elimination through primarily renal plasma clearance processes since the chemotherapeutic is relatively resistant to deamination pathways. | |
| In clinical oncology, fludarabine has been administered for treatment of prostatic adenocarcinoma/carcinoma, ovarian carcinoma, mammary adenocarcinoma/carcinoma, melanoma, and squamous cell carcinoma. The primary indications for the therapeutic administration of fludarabine are for the management of lymphoma [2,3] and leukemia [4-6]. In the treatment of indolent non-Hodgkin’s lymphoma, fludarabine in various combinations is frequently co-administered with cyclophosphamide, mitoxantrone, dexamethasone and Rituximab (anti-CD20). In the FLAG regimen, fludarabine is co-administered in combination with cytarabine and granulocyte colony-stimulating factor (GSF) for treatment of acute myeloid leukemia. Because of its immuno suppression characteristics, fludarabine is also used in some conditioning protocols prior to allogeneic stem cell transplants. Fludarabine is highly effective for the therapeutic management of chronic lymphocytic leukemia (CLL) where it is capable of achieving response rates that are superior to many alkylating agents. Current treatment protocols often induce remission, but prognosis with therapy depends on B-lymphocyte CLL sub-type. Unfortunately, the mortality rate for B-CLL is still relatively high in adults where nearly all cases relapse to an incurable state and all forms of this neoplastic condition have an over-all 5-year survival of only 50% even with immunoglobulin-based therapies (e.g. Rituximab/anti-CD20, Alemtuzumab/anti-CD52). Late-stage B-CLL is frequently treated with several different regiments including chemotherapeutic combinations composed of [i] fludarabine and Rituximab (FR); [ii] fludarabine and cyclophosphamide (FC); [iii] fludarabine, cyclophosphamide, and Rituximab (FCR); and [iv] cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP). The combination of fludarabine and all-trans retinoic acid has enhanced levels of effectiveness against B-CLL [7] as does fludarabine with alkylating agents (cyclophosphamide) which promotes higher response rates and longer progression-free survival. | |
| Despite invaluable applications in modern clinical oncology, fludarabine and many other conventional small-molecular-weight chemotherapeutics almost invariably lack sufficient potency or efficacy as a mono-therapy to completely resolve most advanced or aggressive forms of neoplastic conditions before their administration is limited by the onset of dose-limiting sequelae. Myelo suppression and immuno suppression are the most common and serious toxicities that arise from systemic fludarabine administration. The onset of sequelae is recognized by the detection of anemia, thrombocytopenia and neutropenia so therapeutic intervention routinely requires regular differential white blood cell count monitoring in order to identify conditions requiring supportive administration of whole blood transfusions, platelet-enriched plasma, or G-CSF to stimulate neutrophil production. Fludarabine can also induce profound lymphopenia (e.g. CD4(+) T-lymphocytes) that in turn significantly increases the risk of opportunistic infections. The profound lymphopenia caused by fludarabine increases susceptibility to transfusion-associated graft versus host disease, a fatal complication of blood transfusion. Due to this consideration, it has been advised that only irradiated blood cells be administered post fludarabine therapy. Other significant fludarabine-associated sequelae include the potential to develop severe autoimmune hemolytic anemia (AHA) conditions. | |
| The frequency and degree of risk for the most serious sequelae induced by fludarabine in general increases with elevations in daily dose, total cumulative dose (e.g. high or ultra-high regimens), and duration of therapy. Several molecular strategies can be utilized to minimize the toxic sequelae of fludarabine including their covalent bonding to biologically-relevant molecular platforms that afford properties of selective “targeted” chemotherapeutic delivery [8-12]. The molecular design and an organic chemistry reaction scheme have been developed and implemented in the conception of a multi-phase scheme for the synthesis of a covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic. Lack of anti-IGF-1R fragmentation or IgG-IgG polymerization was established by mass separation using SDS-PAGE analysis in combination with affinity blotting and chemiluminescent autoradiography. Total percent covalent bound chemotherapeutics was determined by measuring the fludarabine concentration contained in the resulting supernatant following protein precipitation in methanol. Retained bindingavidity of the anti-IGF-1R component of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was validated by cell-ELISA. The selectively “targeted” anti-neoplastic cytotoxicity of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] was then determined utilizing populations of pulmonary adenocarcinoma (A549) as an ex-vivo neoplastic disease model. | |
Literature Review |
|
| In contrast to the anthracycline-class of chemotherapeutic agents, organic chemistry reaction regimens for synthesizing covalent fludarabine immuno chemotherapeutics or similar biopharmaceuticals have rarely if ever been described to date. In this context, there are only a limited number of organic chemistry reactions schemes that can potentially be employed to covalently bond fludarabine to biologically-relevant molecular platforms based on its chemical composition and molecular structure. A covalent immunochemotherapeutic can potentially be synthesized through the formation of an amide bond at the fludarabine C6-monoamine group. Analogous organic chemistry reaction schemes have previously been implemented for the synthesis of several covalent epirubicin (C3-monoamine) [8,11] and gemcitabine (C4-monoamine) [12] immuno chemotherapeutics utilizing an amine-reactive N-hydroxysuccinimide ester[8;10-12] or imido-ester covalent bond forming reagent analog [10]. Alternatively, the non-phosphorylated 2-fluoroadenine-9-β-D-arabinofuranoside analog of fludarabine can be covalently bound at the C2-methylhydroxyphosphate group to a biologically-relevant molecular platform utilizing a hydroxylreactive isocyanate reagent as has previously been implemented for the synthesis of gemcitabine-(C5-methylcarbamate)-[anti-HER2/ neu] [9]. | |
| Fludarabine can potentially be covalently bound to a wide array of biologically-relevant molecular platforms. Similar smallmolecular- weight chemotherapeutics have been covalently bonded to poly-L-glutamic acid (polypeptide configuration) [13]; cardiolipin; [14,15]; 1-dodecylthio-2-decyloxypropyl-3-phophatidic acid [16,17]; lipid-nucleosides;[18] N-(2-hydroxypropyl)methacrylamide polymer (HPMA) [19]; benzodiazepine receptor ligands [20,21]; 4-(N)- valeroyl, 4-(N)-lauroyl, 4-(N)-stearoyl [22], and 4-fluoro[18F]- benzaldehyde derivatives (positron emitting radionuclide imaging agent) [23]. Few if any investigations have described the covalent bonding of fludarabine to immunoglobulin, fragments of immunoglobulin (e.g. F (ab’) 2 or Fab’) or receptor ligands (e.g. EGF→EGFR) that can function as large molecular weight platforms that afford properties of selective “targeted” delivery. Endogenous trophic membrane receptors HER2/neu, EGFR, IGF-1R, and VEGFR represent one class of “targets” that can be implemented to selectively “target” the delivery of chemotherapeutic moieties because they are frequently over-expressed by many neoplastic cell types are they are capable of being internalized by active transport mechanisms of IgG/ ligand induced receptor mediated endocytosis [24]. | |
| Several covalent immuno pharmaceutical preparations have been developed that contain moieties that represent alternatives to conventional small-molecular weight chemotherapeutic agents. Many of the anti-cancer moieties in these preparations are relatively expensive and physiologically too toxic to administer systemically as a monotherapy which has motivated their covalent bonding to monoclonal immunoglobulin in the form of [i] colicheamicins which promote DNA strand cleavage (ozogamicin-inotuzumab for CD22(+) lymphoma; ozogamicin-gemtuzumab for CD33(+) leukemia withdrawn 2010); [ii] monomethyl auristatin E (MMAE) tubulin inhibitor (vedotin-glembatumumab for GP-NMP(+) melanoma, glioma, breast cancer; vedotin-brentuximab-vedotin for CD30(+) Hodgkin lymphoma, anaplastic large cell lymphoma); and [iv] maytansines/maytansinoids tubulin inhibitors (emtansine- Trastuzumab for HER2/neu(+) carcinomas; mertansine-bivatuzuma for CD44v6(+) squamous cell carcinoma; mertansine-lorvotuzumab for CD56(+) ovarian carcinoma, small cell lung cancer and multiple myeloma; mertansine-cantuzumab and ravtansine-cantuzumab for Can Ag(+) colorectal cancer). Classical small-molecular weight chemotherapeutics that have become commercially available as covalent immuno chemotherapeutics include; [i] irinotecan metabolite SN38 which inhibits topoisomerase-I (SN38-milatuzumab for CD74(+) multiple myeloma and chronic lymphocytic leukemia/CLL; SN38-labetuzumab for CEA(+) lung, breast and colorectal adenocarcinomas/carcinomas; SN38-hRS7 (IMMU132) for adenocarinomas and carcinomas classified as EPG- 1(+) or TROP-2(+) triple-negative breast cancer, small-cell lung cancer and colorectal cancer); and [ii] anthracyclines which inhibit topoisomerase-II, nucleotide strand formation, “free” oxygen radical formation and chromatin histone eviction (doxorubicin-miltuzumab for CD74(+) chronic lymphocytic leukemia and multiple myeloma). | |
Materials and Methods |
|
| Covalent gemcitabine immunochemotherapeutic synthesis | |
| Phase-I & II synthesis format for amine-reactive chemotherapeutic intermediate: Fludarabine mono-phosphate was formulated at a concentration of 3.85 x 10-2 M in modified PBS buffer (phosphate 5.0 mM, NaCl 75 mM, EDTA 5.0 mM, pH 7.4) and reacted with 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide at a 5:1 molar ratio. The Phase-I & II reaction mixture was then allowed to gently stir at 25ºC for 10 to 15 minutes. | |
| Phase-III synthesis format for covalent fludarabine immuno chemotherapeutics utilizing an amine: reactive chemotherapeutic intermediate - Monoclonal immunoglobulin fractions of anti-IGF- 1R (3.0 mg, 2.0 x 10-5 m moles) devoid of stabilizing reagent was formulated in imidazole buffer (100 mM, pH 6.0) and combined at a 1:50 molar-ratio with the amine-reactive fludarabine- (C2-phosphorylimidazolide) intermediate generated as a end product from the Phase-I & II synthesis reaction scheme. The Phase-III reaction mixture was then gently stirred continuously for 2 hours at 25ºC to maximize the synthesis yield of covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R immunochemotherapeutic end-product. Residual un-reacted (non-covalently bound) fludarabine and reagents were removed from the covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic by serial micro-filtration (MWCO=10-kDa) and buffer exchange utilizing conventional PBS buffer (phosphate 100 mM, NaCl 150 mM, pH 7.4). | |
| Molecular analysis and characterization of properties | |
| Relative non-covalently bound fludarabine content: Detection and monitoring of the relative amount of residual un-reacted (non-covalently bound) fludarabine-C2- methylhydroxyphosphate contained in the Phase-III reaction covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic end-product following serial microfiltration (MWCO=10-kDa) was determined by analytical-scale high-performance thin layer chromatography (HP-TLC silica gel, 250 μm thickness, UV 254 nm indicator). Sensitivity of detecting residual un-reacted fludarabine by analytical HP-TLC was enhanced by evaluation of highly concentrated formulations of the Phase-III covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] immunochemotherapeutic end-product and application of standardized fludarabine/fludarabine-phosphorylimidazolide reference controls formulated at fludarabine-equivalent concentrations. Total fludarabine concentration within the fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] following exhaustive serial micro-filtration was equal to or greater than 10-4 M (36.5 μg/ml) which far exceeds the analytical-scale HPTLC detection limit for fludarabine (100 ng) by several orders of magnitude [25]. Individual analytical-scale HP-TLC silica gel plates were subsequently developed utilizing a mobile phase solvent system composed of propanol/ethanol/ddH20 (17:5:5 v/v ratio). Detection of any residual fludarabine or un-reacted fludarabine-phosphorylimidazolide contained in the Phase-III covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] immunochemotherapeutic following analytical-scale HP-TLC development was determined by direct UV illumination. Complementary methods involve combining the covalent immunochemotherapeutic 1:5 v/v with cold methanol or cold chloroform: isopropanol (2:1 v/v) and measurement of free noncovalently bound chemotherapeutic in the resulting supernatant. | |
| Measurement of covalently bound fludarabine: Total individual absorbance levels at 260-nm were measured for fludarabine-C2-methylhydroxyphosphate, immunoglobulin and immunoglobulin/fludarabine-C2-methylhydroxyphosphate standardized reference controls in addition to the covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic. Concentrations of the immunoglobulin component contained within the Phase-III fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] end-product, and immunoglobulin standardized reference controls were measured at 660-nm utilizing a metal-dye complex reagent (660-nm Protein Assay, Pierce Thermo Scientific). Concentration of the immunoglobulin component within the Phase-III end-product established by measurements at 660-nm were then utilized to calculate the corresponding absorbance measured at 260-nm. Differences between the absorbance for fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] measured at 260-nm and the calculated 260-nm absorbance for the immunoglobulin content of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was utilized to calculate the total fludarabine-equivalent concentration within the Phase-III covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic end-product. | |
| Mass-Separation analysis for detection of polymerization and fragmentation - Covalent fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic in addition to reference control anti-IGF-1R immunoglobulin fractions formulated at a standardized protein concentration of 60 μg/ml were combined 50/50 v/v with conventional SDS-PAGE sample preparation buffer (Tris/glycerol/bromphenyl blue/SDS) without 2-mercaptoethanol or boiling. Covalent fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic, the immunoglobulin reference control (0.9 μg/well) and a mixture of pre-stained molecular weight marker reference controls were then individually developed by non-reducing SDS-PAGE (11% acrylamide) performed at 100 V constant voltage at 3º C for 2.5 hours. | |
| Detection Analyses for Polymerization and Fragmentation- Covalent fludarabine-(C2-methylhydroxyphosphoramide): [anti-IGF-1R] immunochemotherapeutic following mass-separation by non-reducing SDS-PAGE were equilibrated in tank buffer devoid of methanol. Mass/size-separated fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic contained within acrylamide SDS-PAGE gels was then transferred laterally onto sheets of nitrocellulose membrane at 20 volts (constant voltage) for 16 hours at 20C to 30C with the transfer manifold packed in crushed ice. | |
| Covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic laterally transferred onto nitrocellulose membrane was then be equilibrated in Tris buffered saline (TBS: Tris HCl 0.1 M, NaCl 150 mM, pH 7.5, 40 ml) at 40C for 15 minutes followed by an incubation period at 20C to 30C for 16 hours in TBS blocking buffer (Tris 0.1 M, pH 7.4, 40 ml) containing bovine serum albumin (5%) applied in combination with gentle horizontal agitation. Prior to further processing, nitrocellulose membranes were vigorously rinsed in Tris buffered saline (Tris 0.1 M, pH 7.4, 40 ml, n= 3). | |
| Rinsed BSA-blocked nitrocellulose membranes developed for Western-blot (immuno detection) analyses were incubated with HRPO-Protein G conjugate (0.25 μg/ml) at 4ºC for 18 hours on a horizontal orbital shaker. Nitrocellulose membranes following vigorous rinsing in TBS (pH 7.4, 4ºC, 50 ml, n=3) were incubated in blocking buffer (Tris 0.1 M, pH 7.4, with BSA 5%, 40 ml). Blocking buffer was decanted from nitrocellulose membrane blots were vigorously rinsed again in TBS (pH 7.4, 4ºC, 50 ml, n=3) before incubation with HRPO chemiluminescent substrate (25º C; 5-to-10 minutes). Under dark conditions, chemiluminescent autoradiography images were acquired by exposing radiographic film (Kodak Bio Max XAR) to nitrocellulose membranes sealed within transparent ultraclear re-sealable plastic envelops. | |
| Cytotoxic anti-neoplastic potency and ex-vivo neoplastic disease model: | |
| Multi-Drug resistant pulmonary adenocarcinoma tissue culture : The multi-drug resistant human pulmonary adenocarcinoma/alveolar basal epithelial cell line (A549 derived in 1972 from a 58 year old Caucasian male) was utilized as an ex-vivo model for neoplastic disease. Pulmonary adenocarcinoma (A549) populations were propagated in 150-cc2 tissue culture flasks containing F-12K growth media supplemented with fetal bovine serum (10% v/v) and penicillinstreptomycin at a temperature of 37º C under a gas atmosphere of carbon dioxide (5% CO2) and air (95%). Growth media was not supplemented with growth factors, growth hormones or any other type of growth stimulant. Trypsin or any other biochemically active enzyme fractions were not used to facilitate harvest of pulmonary adenocarcinoma (A549) cell suspensions for seeding of tissue culture flasks or multi-well tissue culture plates. Pulmonary adenocarcinoma (A549) monolayer populations utilized for cell-ELISA analyses were uniformly propagated to a >85% level of confluency. | |
| Cell-ELISA total membrane-bound immunoglobulin assay: Pulmonary adenocarcinoma (A549) cell suspensions were seeded into 96-well microtiter plates in aliquots of 2×105 cells/well and allowed to form a confluent adherent monolayer over a period of 24- to-48 hours. The growth media content in each individual well was removed manually by pipette and the cellular monolayers were then serially rinsed (n= 3) with PBS followed by their stabilization onto the plastic surface of 96-well microtiter plates with paraformaldehyde (0.4% in PBS, 15 minutes). Stabilized cellular monolayers were then incubated in triplicate with gradient concentrations of covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic formulated at immunoglobulinequivalent concentrations of 0.01, 0.10, 1.00 and 10.00 μg/ml in tissue culture growth media (200 μl/well). Direct contact incubation between pulmonary adenocarinoma (A549) monolayers and fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was performed at 370C over a 3-hour incubation period under a gas atmosphere of carbon dioxide (5% CO2) and air (95%). Following serial rinsing with PBS (n=3), development of stabilized pulmonary adenocarcinoma (A549) monolayers entailed incubation with β-galactosidase conjugated goat anti-mouse IgG (1:500 dilution) for 2 hours at 25ºC with residual unbound immunoglobulin removed by serial rinsing with PBS (n=3). Final development of the cell-ELISA required serial rinsing (n=3) of stabilized pulmonary adenocarcinoma (A549) monolayers with PBS followed by incubation with nitrophenyl-β-D-galactopyranoside substrate (100 μl/well of ONPG formulated fresh at 0.9 mg/ml in PBS pH 7.2 containing MgCl2 10 mM, and 2-mercaptoethanol 0.1 M). Absorbance within each individual well was measured at 410 nm (630 nm reference wavelength) following incubation at 37°C for a period of 15 minutes. | |
| Cell proliferation-vitality assay for measuring cytotoxic antineoplastic potency: Individual preparations of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] were formulated in growth media at final standardized fludarabine-equivalent concentrations of 10-10, 10-9, 10-8, 10-7, and 10-6 M. Each standardized fludarabine-equivalent concentration of the covalent immuno chemotherapeutics was then transferred in triplicate into 96-well microtiter plates containing adherent pulmonary adenocarcinoma (A549: 2000 cells/well) and growth media (200 μl/well). Covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic was then incubated in direct contact with pulmonary adenocarcinoma (A549) monolayer populations for a period of 192-hours (37°C under a gas atmosphere of carbon dioxide (CO2 5%) and air (95%). Following the initial 96-hour incubation period, pulmonary adenocarcinoma (A549) populations were replenished with fresh tissue culture media with or without covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic. | |
| Anti-neoplastic cytotoxic potency of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] was measured by removing all contents within the 96-well microtiter plates manually by pipette followed by serial rinsing of stabilized monolayers (n=3) with PBS followed by incubation with 3-[4,5-dimethylthiazol-2-yl]- 2,5-diphenyl tetrazolium bromide vitality stain reagent formulated in RPMI-1640 growth media devoid of pH indicator or bovine fetal calf serum (MTT: 5 mg/ml). During an incubation period of 3-4 hours at 37OC under a gas atmosphere of carbon dioxide (5% CO2) and air (95%) the enzyme mitochondrial succinate dehydrogenase was allowed to convert the MTT vitality stain reagent to navy-blue formazone crystals within the cytosol of pulmonary adenocarcinoma (A549) cell populations (some reports suggest that NADH/NADPH dependent cellular oxidoreductase enzymes may also be involved in the biochemical conversion process). Contents were then removed from each of the 96-wells in the microtiter plate, followed by serial rinsing with PBS (n=3). The resulting blue intracellular formazone crystals were dissolved with DMSO (300 μl/well) and then spectro photometric absorbance of the resulting blue-colored supernatant measured at 570 nm using a computer-integrated microtiter plate reader. | |
Results |
|
| Covalently-bound fludarabine content: The predominant Phase-I end-product in PBS at pH 7.4 is a reactive fludarabine carbodiimide phosphate ester intermediate complex (Figure 1). Addition of the reactive fludarabine carbodiimide phosphate ester intermediate to immunoglobulin formulated in imidazole buffer at pH 6.0 preferentially produces a transient Phase-II amine reactive fludarabine-phosphorylimidazolide intermediate (Figure 1). The aliphatic ε-monoamine of the lysine amino acid side chain then preferentially reacts with the Phase-II fludarabine phosphorylimidazole intermediate because of much stronger base characteristics compared to aromatic amines such as those found at the C6 position of fludarabine and in aniline due to the electron sink effect from the associated organic ring structures (Figure 1). The covalent phosphoramide bond structure is highly stable at 4°C or in whole plasma or tissue culture media like environments containing 5% plasma or 5% serum albumin [27] in contrast to strictly aqueous buffer solutions devoid of biological proteins where at 37°C approximately a 12% total liberation rate occurs over a 100 hour period [28]. | |
| Figure 1: Organic Chemistry Reaction Scheme for Synthesis ofCovalent Fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] Immunochemotherapeutic. Phase-I Reaction Scheme(Row Plate ?): reaction of the fludarabine C2-mono-phosphategroup with 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide totransiently form a reactive fludarabine carbodiimide phosphateester intermediate complex; Phase-II Reaction Scheme (Row Plate??): rapid spontaneous conversion of the transient Phase I reactiveintermediate to the Phase-II fludarabine-phosphorylimidazolideamine-reactive intermediate in the presence of imidazole. Phase-IIlReaction Scheme (Row Plate ???): covalent phosphoramide bondformation between the Phase-ll fludarabine-phosphorylimidazolideamine-reactive intermediate and µ-mononamine of lysine residueswithin the amino acid sequence of anti-IGF-1R monoclonalimmunoglobulin resulting in the synthesis of a covalentfludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]immunochemotherapeutic. | |
| Molar-incorporation index: The calculated fludarabine IgG molar-incorporation-index for covalent fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was 3.67:1 utilizing the organic chemistry reaction scheme to form a covalent phosphoramide bond at the C2-methylhydroxyphosphate group of fludarabine (Figure 1). Micro-filtration (MWCO=10-kDa) provided substantially greater yield levels for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] than did the removal of residual chemotherapeutic and un-reacted chemical reagents by micro-scale size exclusion column chromatography. | |
| Mass-Separation Analysis for Detection of Polymerization and Fragmentation- Molecular weight profile analysis of covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immuno chemotherapeutics mass-separated by SDS-PAGE in combination with immunodetection analysis (Western-blot) and chemiluminescent autoradiography recognized a single primary condensed band of 150-kDa between a molecular weight range of 5.0- kDa to 450-kDa (Figure 2). Profiles consistent with low-molecularweight fragmentation (proteolytic/hydrolytic degradation) or largemolecular- weight IgG-IgG polymerization were not detected (Figure 2). The observed molecular weight of 150-kDa for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] directly corresponds with the known molecular weight/mass of reference control anti-IGF-1R monoclonal immunoglobulin fractions (Figure 2). Analogous results have been reported for similar covalent immuno chemotherapeutics [8-12,29,30]. | |
| Figure 2: Characterization of the molecular weight profile for thecovalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] immunochemotherapeutic relative to reference controlanti-IGF-1R monoclonal immunoglobulin fractions and proteinmolecular weight standards. Legends: (Lane-1) murine anti-humanEGFR monoclonal immunoglobulin; and (Lane-2) fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]. The covalentfludarabine immunochemotherapeutic and monoclonalimmunoglobulin fractions were size-separated by non-reducingSDS-PAGE followed by lateral transfer onto sheets of nitrocellulosemembrane to facilitate detection with HRPO-Protein G conjugate.Subsequent analysis entailed incubation with a HRPOchemiluminescent substrate and the acquisition of autoradiographyimages. | |
| Figure 3: Evaluation of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] by analytical HPTLCfor the detection of residual fludarabine not covalently boundto anti-IGF-1R immunoglobulin. (Lane-1) Phase-II fludarabinephosphorylimidazolideamine-reactive intermediate; and (Lane-2)Phase-III covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]immunochemotherapeutic following serial micro-filtration(MWCO = 10-kDa). Standardized fludarabine-equivalentconcentrations of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] and the fludarabine-phosphorylimidazolide aminereactiveintermediate were applied to HP-TLC plates (silica gel, 250µm thickness, UV 254 nm indicator) and developed utilizing apropanol/ethanol/H20 (17:5:5 v/v) mobile phase. Identification ofany residual fludarabine or un-reacted fludarabinephosphorylimidazolidein the Phase-III covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]immunochemotherapeutic was subsequently determined by directillumination with UV light. | |
| Cell-ELISA total membrane immunoglobulin binding analysis: Total immunoglobulin in the form of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] bound on the external surface membrane of adherent pulmonary adenocarcinoma (A549) monolayer populations was detected and measured by cell-ELISA (Figure 4). | |
| Figure 4: Detection of total immunoglobulin in the form offludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]selectively bound to the exterior surface membrane of pulmonaryadenocarcinoma. Covalent fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]immunochemotherapeutic formulated at gradientimmunoglobulin-equivalent concentrations were incubated indirect contact with triplicate monolayer populations ofchemotherapeutic-resistant human pulmonary adenocarcinoma(A549) over a 4-hour time period. Total immunoglobulin bound tothe exterior surface membrane was then detected and measured bycell-ELISA. | |
| Increases in fludarabine-(C2- methylhydroxyphosphoramide)- [anti-IGF-1R] formulated at the standardized immunoglobulinequivalent concentrations of 0.010, 0.10, 1.00 and 10.00 μg/ml corresponded with progressive elevations in the total amount of membrane-bound immunoglobulin (Figure 4). Collectively results from cell-ELISA analyses validated the retained selective bindingavidity of fludarabine-(C2- methylhydroxyphosphoramide)-[anti- IGF-1R] for external membrane IGF-1R receptor sites highly over-expressed on the exterior surface membrane of pulmonary adenocarcinoma (A549) monolayer populations (Figure 4). | |
| Cytotoxic Anti-Neoplastic Potency - Nearly identical levels of cytotoxic anti-neoplastic potency were detected individually for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF- 1R] and fludarabine against pulmonary adenocarcinoma (A549) when challenged with fludarabine-equivalent concentrations at and between 10-9 M -to- 10-5 M over a 192-hour incubation period (Figure 5). Anti-neoplastic cytotoxicity of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] gradually increased at and between the standardized fludarabine-equivalent concentrations of 10-9 M, 10-8 M, 10-7 M and 10-6 M which corresponded with lethal cancer cell death percentages of 6.2%, 19.7%, 20.9%, and 24.4% (93.8%, 80.3%, 79.1% and 75.6% residual survival) respectively (Figure 5). A dramatic elevation in anti-neoplastic cytotoxicity for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was detected at and between the standardized fludarabine-equivalent concentrations of 10-6 M and 10-5 M where lethal cancer cell death percentage levels increased from 24.4% to a maximum of 94.7% (75.6% to 5.3% residual survival) respectively (Figure 5). | |
| Figure 5: Relative cytotoxic anti-neoplastic potency of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] againstchemotherapeutic-resistant pulmonary adenocarcinoma. Legends:Formulated in triplicate at gradient standardized (fludarabineequivalent)concentrations, both fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] and fludarabinewere separately incubated in direct contact with monolayerpopulations of chemotherapeutic-resistant pulmonaryadenocarcinoma (A549) for a period of 192-hours. Cytotoxic antineoplasticpotency was measured using a MTT cell vitality assayrelative to matched negative reference controls. | |
Discussion |
|
| The molecular design and the organic chemistry reaction scheme implemented in the development of the multi-phase synthesis regimen for fludarabine-(C2- methylhydroxyphosphoramide)-[anti- IGF-1R] generates a covalent phosphoramide bond structure at the C2-methylhydroxyphosphate group of fludarabine (Figure 1) [31,32]. The organic chemistry reactions described for the synthesis of the covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic represents a departure from previously described methodologies for covalently bonding small-molecular-weight chemotherapeutics to biologicallyrelevant molecular platforms. Specifically, the multi-phase organic chemistry reaction scheme for the synthesis of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] initially generates a transient and reactive Phase-I fludarabine carbodiimide phosphate ester intermediate which in the presence of imidazole is rapidly transformed into a more stable Phase-II fludarabine amine-reactive phophorylimidazole intermediate (Figure 1). Formulation of fludarabine at a relatively large molar excess to the carbodiimide reagent served to promote maximal yield of the Phase II amine-reactive fludarabine intermediate; maximize depletion of the carbodiimide reagent (also unstable for prolong incubation periods in aqueous buffer systems); accelerate the rate of the organic chemistry reaction; and minimize the risk of large molecular weight IgG-IgG polymerization. The more stable Phase-II reaction end-product if synthesized in an anhydrous solvent system can potentially be preserved as an aminereactive fludarabine phophorylimidazole intermediate thereby providing a degree of flexibility and practicality that allows the option of covalently bonding the chemotherapeutic moiety to a variety of different molecular platforms that contain available primary amine groups. The Phase-II fludarabine phophorylimidazole intermediate was ultimately reacted with the ε monoamine group of lysine residues within the amino acid sequence of anti-IGF-1R monoclonal immunoglobulin resulting in the formation of a methylhydroxyphosphoramide bond structure at the C2 position and production of the Phase-III fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] end-product (Figure 1). | |
| Efficiency and effectiveness of the multi-phase organic chemistry reaction regimen implemented for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] is at least partially illustrated by the 3.67:1 fludarabine: IgG molar-incorporation-index attained during the course of the synthesis scheme. Such a perspective is further substantiated by experimental results from analogous synthesis investigations where 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide in combination with imidazole can be used to covalently bond other phosphorylated pharmaceutical analogs to monoclonal immunoglobulin fractions (research results pending publication). The fludarabine: IgG 3.67:1 molar-incorporationindex for fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] was comparatively greater than values obtained in previous investigations that employed other covalent bond forming agents for synthesizing; [i] gemcitabine-(C5-carbamate)-[anti-HER2/neu] (Gem: IgG=1.1-to-1) [9] [ii] gemcitabine-(C4-amide)-[anti-HER2/ neu] (Gem: IgG=2.78-to-1);[12] [iii] epirubicin-(C3-amide)-[anti HER2/neu] (Epi:IgG=0.275-to-1); [8] [iv] epirubicin-(C3-amide)- [anti-EGFR] (Epi:IgG=0.407-to-1); [8] and [vi] epirubicin-(C13- imino)-[anti-HER2/neu] (Epi:IgG=0.400-to-1) [10]. | |
| The classic variables of temperature, concentration and reaction time duration can be modified to significantly enhance the efficiency and yield of organic chemistry reactions. Other conditions and parameters likely contributed to achieving the fludarabine: IgG molar-incorporation-index of 3.67:1 for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] such as the; [i] lack of a pre-thiolation requirement for anti-IGF-1R immunoglobulin; [ii] potentially greater chemical reactivity of carbodiimide analogs compared to other covalent bond forming reagents; [iii] enhanced preferential phosphate reactivity of 1-ethyl-3-[3- dimethylaminopropyl]carbodiimide in the presence of imidazole; [iv] formation of a covalent bond with an available phosphate group at the C2-methylhydroxyphosphate position in contrast to a phosphate (Ar-PO4 -), carboxyl (Ar-CO2 -), amine (Ar-NH3 +) or sulfhydryl (Ar- SH) chemical group located directly on an aromatic ring structure; [v] restricting the initial chemical reaction of the carbodiimide reagent with only fludarabine-C2-monophosphate in a manner that enhanced yield and minimized generation of side reaction endproducts (IgG-based determination); [vi] formulation of fludarabine- C2-methylhydroxyphosphoramide in molar excess to the carbodiimide reagent in order to promote and maximize its depletion; [vii] selective amine reactivity of the Phase-II fludarabine-(C2-methylhydroxyph ophorylimidazole) intermediate; and [viii] presence of only a single phosphate group within the chemical composition of fludarabine- C2-methylhydroxyphosphate. Availability of the monophosphate group at the C2-methylhydroxyphosphate position reduces both the degree of steric-hinderance phenomenon in the application of most carbodiimide reagents, while also decreasing unique influences from aromatic ring electron orbitals that often modify the properties of phosphate (Ar-PO4 -), carboxyl (Ar-CO2 -), amine (Ar-NH3 +) or sulfhydryl (Ar-SH) groups. Complementing the effectiveness of the organic chemistry reactions, the final yield of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] was substantially improved by implementing both a multi-phase organic chemistry reaction scheme (in preference to a single-phase “co-mingled” regimen) in concert with serial micro-filtrations (MWCO 10-kDa) instead of micro-scale column chromatography for separating and purifying the Phase-III covalent immunochemotherapeutic endproduct. | |
| Logistically, the organic chemistry reactions implemented in the multi-phase synthesis regimen for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] have distinct attributes as an alternative to other methods described for production of covalent immunochemotherapeutics. Most notable in this regard is the comparatively rapid in reaction duration compared to methods that employ dicyclohexylcarbodiimide; relatively high yield of covalent immunotherapeutic end-product (IgG-based determination), low to moderately low level of technical difficulty; and marginal dependence on access to advanced forms of laboratory instrumentation. The organic chemistry reaction scheme in the multi-phase synthesis regimen also allows the option of producing an amine-reactive Phase-II fludarabine intermediate that is sufficiently stable for short-to-long-term preservation/storage, or substituting other chemotherapeutic agents or pharmaceuticals for fludarabine, or substituting other biologically-relevant molecular platforms for anti-IGF-1R monoclonal immunoglobulin. | |
| The covalent fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] immunochemotherapeutic possesses several distinct characteristics from the perspective of chemical composition and molecular structure that are important to recognize. Desirable characteristics in this regard are; [i] a specific activity greater than 1:1 (fludarabine: IgG molar-incorporation-index=3.67:1); [ii] generation of C2-methylhydroxyphosphoramide while covalently bonding fludarabine to anti-IGF-1R which at least theoretically provides a higher level of fludarabine moiety bioavailability within the acidic micro-environment of the phagolysosome following internalization by active transport mechanisms of selective “targeted” IgG-induced receptor-mediated endocytosis; [iii] retained biological activity as a function of binding-avidity for over-expressed IGF-1R (detected by cell-ELISA); [iv] in-vivo capacity to induce ADCC, complementmediated lysis and opsonization/phagocytosis; and [v] no insertion or addition of artificial or foreign chemical groups during the synthetic formation of the C2-methylhydroxyphosphoramide bond structure which in turn decreases the risk of inducing host humoral immune responses. | |
| In addition to the potential for fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] to function as an effective anti-cancer agent, it’s chemical composition and molecular configuration along with the multi-phase organic chemistry reactions implemented for development of the multi-phase synthesis scheme can serve as a reference prototype or template for guiding the molecular design and synthesis of other future covalent immunochemotherapeutics. Relevant examples in this regard are the general molecular structure and organic chemistry reaction schemes that can be a component of multi-phase synthesis schemes for synthesis of covalent immunochemotherapeutics analogous to decitabine-(C2-methylhydroxyphosphoramide)-[anti-CD19], clofarabine-(C2-methylhydroxyphosphoramide)-[anti-CD20], and cytarabine-(C2-methylhydroxyphosphoramide)-[anti-CD52], cladribine-(C2-methylhydroxyphosphoramide)-[anti-CD19], or 5-azacitidine-(C2-methylhydroxyphosphoramide)-[anti-CD20] that employ related phosphorylated chemotherapeutics as moieties. Covalent immunochemotherapeutics of this type would possess properties of selective “targeted” delivery and have the expected potential of being able to exert anti-neoplastic cytotoxicity against various forms of leukemia and lymphoma [33-35]. | |
| In the selection of molecular platforms that chemotherapeutics or other pharmaceuticals will be covalently bonded to for the purpose of imparting properties of selective “targeted” delivery, such entities should ideally possess one or preferably more important biological characteristics in the form of; [i] binding-avidity specifically restricted to a unique or highly over-expressed entity on the exterior surface membrane of (neoplastic) cell populations; [ii] bindingavidity that blocks physical interactions between endogenous ligands and their corresponding trophic membrane receptors in a manner that suppresses neoplastic cell vitality and proliferation; [iii] binding-avidity that inhibits the function of trophic membrane receptors by inducing transient or permanent declines in surface membrane expression through mechanisms of IgG-induced receptor mediated endocytosis; and/or the [iv] ability to induce or initiate internalization of covalent immunochemotherapeutics or covalent ligand-chemotherapeutics by active transport mechanisms of receptor-mediated-endocytosis. Other desirable attributes of molecular platforms applied to facilitate properties of selective “targeted” delivery as associated with the chemical groups they contain that can potentially be utilized to create synthetic covalent bond structures. Optimum results are attainable when amine (-NH3 +), and less frequently carboxyl (CO2 -), hydroxyl (-OH) or sulfhydryl (-SH) chemical groups are [v] relatively abundant (e.g. most protein sulfhydryl R-SH groups are not abundant); [vi] physically available (minimal steric-hinderance phenomenon); [vii] possess a molecular weight/size sufficiently large enough (e.g. >60-kDa) to prolong the plasma pharmacokinetic profile of the chemotherapeutic moiety through delayed or prevented excretion by renal plasma clearance; and [viii] ability in-vivo to stimulate endogenous immune responses of antibody-dependent cell cytotoxicity (ADCC), complementmediated cytolysis, and opsonization/phagocytosis. | |
| Significant advances have been made in identifying trophic membrane receptors over-expression in many adenocarcinoma and carcinoma affecting the breast, prostate, intestine, ovary or kidney and the implications of this phenomenon on cancer cell biological phenomenon [36] as it pertains to viability [37,38] proliferation rate [38,39], local invasiveness [40] metastatic potential [41,42] and chemotherapeutic resistance (e.g. P-glycoprotein co-expression) [40,43,44]. Anti-trophic receptor monoclonal immunoglobulins that bind to and inhibit the function of trophic receptor complexes uniquely or highly over-expressed on the external surface membrane of many neoplastic cell types independently suppress growth rate and vitality through several mechanisms. Suppression of the biological function of trophic membrane receptors therefore can occur through simple competitive inhibition or “blocking” of endogenous ligand binding (e.g. EGF→EGFR) that in turn may also be accompanied by transient decreases in surface membrane expression density that occur secondary to active transport mechanisms of IgG/ligand induced receptor-mediated endocytosis. Additionally, but variable degrees of selective “targeted” anti-neoplastic cytotoxicity can be attained in-vivo with anti-trophic receptor monoclonal immunoglobulin through stimulation of host immune responses by the formation of membrane Ag: IgG complexes that activate; [i] antibody-dependent cell cytotoxicity (ADCC); [ii] complement-mediated cytolysis, and [iii] optimization/phagocytosis. Anti-HER2/neu (Trastuzumab, Pertuzumab) [45-49], anti-EGFR (Cetuximab) [50-53], combined anti-HER2/neu and anti-EGFR (Panitumumab) [52-55], and anti- IGF-1R (Figitumumab, Dalotuzumab) [56-59] represent some of the anti-trophic receptor monoclonal immunoglobulins most extensively utilized in clinical oncology for the therapeutic management of adenocarcinomas and carcinomas affecting the breast, prostate, intestine, kidney and lung. | |
| Leukemia and lymphoma cell types most frequently do not uniquely or highly over-express classical endogenous receptor complexes on their external surface membrane as do many nonhaemopoietic cancer cell types. Many leukemia and lymphoma cell types, however, do frequently highly over-express or uniquely express several membrane cell differentiation antigens such as CD20, CD22, CD33 (SIGLEC: sialic acid binding lectin), and CD54. Each of these cell differentiation antigens is the basis for the therapeutic monoclonal immunoglobulins [i] anti-CD20 (Rituximab: non-Hodgkin’s lymphoma; Ofatumumab: chronic lymphocytic leukemia; Veltuzumab: non-Hodkin lymphoma; Trubion: chronic lymphocytic leukemia and non-Hodgkin lymphoma); and [ii] anti-CD52 (Alemtuzumab: chronic lymphocytic leukemia/CLL). In contrast to the anti-trophic receptor immunoglobulins that suppress the growth and vitality of non-haemopoietic neoplastic cell types like the adenocarcinomas and carcinomas, the cytotoxic anti-neoplastic properties of anti-CD20 and anti-CD54 attained in-vivo against populations of leukemia and lymphoma cell types is highly dependent upon if not largely confined to the activation of endogenous immune responses [60-69]. Majority if not all of the in-vivo anti-neoplastic cytotoxic properties of anti-CD20, anti- CD33 and anti-CD54 is therefore predominately associated with their ability to induce antibody-dependent cell cytotoxicity (ADCC), complement-mediated cytolysis, and opsonization/phagocytosis. Monoclonal anti-CD22 immunoglobulin has similarly been applied in the development of both inotuzumab-ozogamicin (possess a highly toxic calicheamicin as a functional chemotherapeutic moiety) and Moxetumomab pasudotox (CAT-8015: pseudomonas-origin exotoxin moiety). Each of these anti-CD22 based biotherapeutics have demonstrated efficacy against non-Hodgkin lymphoma or hairy cell leukemia respectively. The monoclonal anti-CD33 immunoglobulinbased immunochemotherapeutics, gemtuzumab-ozogamicin (2000- to-2010 withdrawal), and SGN-CD33 are covalently bound to a pyrrolobenzodiazepine chemotherapeutic moiety (promotes intrastrand DNA cross-linking) which has been variably effective in the therapeutic management of acute myeloid leukemia. Some of the anticancer properties attained with anti-CD33 therapeutic monoclonal immunoglobulins or anti-CD33 covalent immunochemotherapeutics are associated with the tyrosine-based inhibitory motif located intracellularly and its efficacy or influence on inhibiting cellular activity. Similarly, monoclonal anti-CD56 immunoglobulin covalently bound to mertansine (maytansinoid analog) in the form of Lorvotuzumabmertansine for the treatment of multiple myeloma. Related covalent mertasine analogs include Bivatuzumab-mertansine, Cantuzumabmertansine and Trastuzumab-emtansine that can be effective against metastatic head and neck cancer (anti-CD44), colorectal cancer (anti-CanAg) and Trastuzumab-emtansine (T-DMI anti-HER2/neu positive adenocarcinomas/carcinomas) respectively. | |
| Therapeutic monoclonal immunoglobulin can potentially suppress neoplastic cell vitality though mechanisms-of-action that are distinctly different from those associated with conventional smallmolecular- weight chemotherapeutic agents. The biological function and characteristics of anti-IGF-1R are very similar to the properties of anti-HER2/neu and anti-EGFR preparations. Monoclonal immunoglobulin with IGF-1R binding-avidity competitively disrupts IGF ligand binding and stimulation resulting in declines in survival and proliferation of adenocarcinomas and carcinomas affecting the lung, breast and prostate in addition to some sarcomas. Other biological functions of cancer populations regulated by IGF-1R overexpression that are inhibited by anti-IGF-1R include neoplastic cell transformation, mobility/migration and metastatic potential. Such qualities suppress neoplastic cell sub-populations refractory (resistant) to conventional small-molecular-weight chemotherapeutics while at the same time avoiding the risk of many serious sequellae. | |
| Despite the potential for anti-IGF-1R, anti-HER2/neu, anti- EGFR, anti-IGF-1R and similar monoclonal immunoglobulin-based modalities to inhibit growth and proliferation of neoplastic cell populations, they are unfortunately almost all invariably plagued by an inability to evoke cytotoxic activity sufficient to independently resolve most aggressive or advanced forms of neoplastic diseases as a monotherapy [45,46,70-84]. Inability of most anti-trophic receptor immunoglobulins to exert significant cytotoxic efficacy in-vivo is in part based on detection of increases in cell-cycle G1- arrest, cellular transformation to states of apoptosis-resistance [71] and increased expansion of resistant sub-populations [45,46] which can be further complicated by frequent reversal of tumor growth inhibition [45] and resumption of trophic receptor over-expression [70] following cessation of immunoglobulin pressure. Greater anti-neoplastic cytotoxicity can alternatively be attainable when an | |
| anti-trophic receptor immunoglobulin are utilized in concert with conventional chemotherapeutics or other anti-cancer modalities [85- 87]. In this context, additive and synergistic levels of anti-neoplastic properties are possible with anti-HER2/neu in dual combination with cyclophosphamide [86,88], docetaxel [88], doxorubicin [86,88], etoposide [88], methotrexate [88], paclitaxel [86,88], or vinblastine [88]. Similar to anti-HER2/neu [86,88-92], other trophic receptor site inhibitors including anti-EGFR [93-95], anti-IGFR-1 [96,97], and anti-VEGFR [85,98,99] also produce additive and synergistic levels of anti-neoplastic cytotoxicity when applied in combination with conventional small molecular weight chemotherapeutics. Therapeutic resistance has been found to develop occasionally with anti-CD20 (Veltuzumab, Ofatumumab) and anti-CD52 (Alemtuzumab) through active transport mechanisms that involve accelerated rates of receptor-mediated-endocytosis prior to ADCC/CMC/opsonization [60,100], and monocyte/macrophage CD20/CD52 “shaving” (trogocytosis) [61]. Interestingly, fatumumab has been approved for B- CLL resistant to Alemtuzumab and fludarabine. | |
| In most scenarios, monoclonal immunoglobulin with bindingavidity for over-expressed trophic membrane receptors (e.g. IGF-1R, EGFR or HER2/neu) evoke no or very limited degrees of selective anti-neoplastic cytotoxicity in the form of suppressed cancer cell proliferation and vitality in an ex-vivo tissue culture environment [8,30,101-104] in contrast to their in-vivo therapeutic properties. Multiple reasons contribute to this observation in part due to the same reasons why same anti-trophic receptor monoclonal immunoglobulins do not as a monotherapy exert sufficient levels of anti-neoplastic cytotoxicity. In addition, the time period of evaluation is relatively brief, growth media utilized in tissue culture environments by convention contain only 5% to 10% v/v bovine serum that is not supplemented with endogenous human receptor ligand, and the host immune responses of ADCC, complement mediated cytolysis and opsonization do not present themselves. | |
| In spite of therapeutic limitations associated with anti-trophic receptor monoclonal immunoglobulin, their binding-avidity for uniquely or highly over-expressed endogenous receptors on the external surface membrane of many neoplastic cell types makes them ideally suited as the key component of covalent immunochemotherapeutics that facilitates properties of selectively “targeted” chemotherapeutic delivery. Several cancer cell biology variables directly influence the selective “targeted” delivery and anti-neoplastic cytotoxic potency of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] and other analogous covalent immunochemotherapeutics [8-12]. Absolute uniqueness of membrane expression determines how selectively “targeted” the chemotherapeutic moiety is delivered. Conversely, the density of membrane expression and the rate at which these same sites are replenished following internalization by mechanisms of receptor-mediated endocytosis govern the fraction of dose deposited, accumulated cytosol concentration and ultimately anti-neoplastic cytotoxic potency. In clinical oncology, the basic principles of pharmacology imply that the property of selective “targeted” chemotherapeutic delivery in general is a more valuable quality than potency if the margin-of-safety is high because therapeutic dosage can be adjusted. | |
| In direct accord with the inter-dependent relationship between the immunoglobulin component of covalent immunochemotherapeutics, and the cell biology characteristics of individual neoplastic cell types, there are several other variables in addition to the expression density for membrane-associated “target” sites that likely contribute significantly to the anti-neoplastic cytotoxicity of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] and similar covalent immunochemotherapeutic agents [8-12]. Intracellular internalization of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] by active transport mechanisms of receptor-mediated endocytosis [24] induced following binding of immunoglobulin or other ligand component (e.g. gemcitabine-EGF) minimizes or avoids a simple “coating” of a covalent immunochemotherapeutic on the external surface membrane of neoplastic cells. Importance of this process is based on the general prerequisite for most classical chemotherapeutic agents like fludarabine to be transported across the lipid bilayer membrane of neoplastic cells due to their mechanisms-of-action which are usually dependent upon their entry into cytosol or nuclear environments in order to create a biological effect. Similar transport processes are assumed to not be required for anti-cancer therapeutics that are membrane-active agents or radioimmunopharmaceuticals that have mechanisms-of-action that do not require entry into the cytosol or nuclear environments (e.g. [213Bi or 211At or 224Ra]-anti- TAG-72 for colon carcinoma). | |
| Covalent bonding fludarabine to a large molecular weight platform possessing binding-avidity for a membrane-associated receptor or antigen that is uniquely or highly over-expressed by a neoplastic cell type can provide an opportunity for simultaneously facilitating the selective “targeted” delivery of chemotherapeutic moieties in addition to a molecular mechanism for maximizing therapeutic efficacy and potency. In instances when a monoclonal immunoglobulin is utilized to produce a covalent immunochemotherapeutic like fludarabine- (C2-methylhydroxyphosphoramide)-[anti-IGF-1R] which possesses binding-avidity for an endogenous membrane receptor known to be internalized by active transport mechanisms of IgG or ligand induced receptor-mediated endocytosis then it becomes possible to also elevate levels of active trans-membrane transport to the extent that it promotes substantial cytosol accumulation of a chemotherapeutic moiety. Attributes that are a consequence of this biological phenomenon in neoplastic populations is the capacity to increase intracellular chemotherapeutic concentrations to levels that are 8.5x [105] to >100x [106,107] higher than those safely attained with conventional small molecular weight chemotherapeutics by simple passive diffusion from the extracellular fluid compartment following intravenous injection at clinically relevant dosages. Although specific data for IGF-1R receptor-mediated endocytosis is somewhat limited for pulmonary adenocarcinoma (A549), other neoplastic cell types like Lewis Lung carcinoma (H-59: highly metastatic subline with a hepatic propensity), and mammary adenocarcinoma (MCF-7) are known to internalize membrane IGF-1R receptors by the active transport mechanism of receptor-mediated endocytosis at a rate of ≈2.1×104/cell (54%) and ≈ 4.5×104/cell (45%) within a 1-hour IGF incubation period [108]. Related investigations have demonstrated that metastatic multiple myeloma internalizes and metabolizes approximately 8×106 molecules of anti-CD74 monoclonal antibody per day [109]. Given this perspective, one of the most critically important cancer cell biology characteristics that determines the cytotoxic anti-neoplastic potency of covalent immunochemotherapeutics like fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R], gemcitabine-(C5-carbamate)-[anti-HER2/neu] [9], gemcitabine-(C4- amide)-[anti-HER2/neu] [12], epirubicin-(C3-amide)-[anti-HER2/ neu][8], epirubicin-(C3-amide)-[anti-EGFR] [8], epirubicin-(C13- imino)-[anti-HER2/neu][10] is the expression density for external membrane-associated trophic receptor utilized as “targets” relative to normal tissues and organ systems. Ideally, in the process of identifying a site on the external surface membrane for the purpose of facilitating selective “targeted” chemotherapeutic delivery, it is there for a distinct advantage for them to not only be present at high expression density levels, but also be capable of being internalized by inducible active transport mechanisms similar to IgG or endogenous ligand initiated receptor-mediated endocytosis. Furthermore, it is vitally important that sites located on the external membrane be chosen so that they can facilitate not only selective “targeted” chemotherapeutic delivery, but are also known to functionally undergo phenomena analogous to receptor-mediated-endocytosis in order to avoid simple “coating” of the external surface of cancer cell membranes. Such a prerequisite is relevant assuming that the anti-cancer agent moiety has a mechanismof- action that is dependent upon an ability to modify the function of molecular entities within the cytosol or nuclear environments in order to exert a biological effect. Such a requirement would not be a prerequisite for anti-cancer agents that instead alter or disrupt the physical integrity of cancer cell membranes or the function of complexes that are an integral component of membrane structures (e.g. ricin, photodynamic agents). | |
| The cytotoxic anti-neoplastic potency of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] against pulmonary adenomacarcinoma (A549) was nearly identical to fludarabine when formulated at and between the standardized fludarabine-equivalent concentrations of 10-9 M and 10-6 M (Figure 4). Based on the molecular weight of fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] and the known mechanism-of-action for fludarabine, these results validate the perspective that the covalent fludarabine immunochemotherapeutic was effectively internalized by active transport mechanisms of receptor-mediated endocytosis following selective “targeted” binding at over-expressed IGF-1R receptors. Because of the relatively common administration of fludarabine for the treatment of chronic lymphocytic leukemia (CLL), fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] represents a prototype covalent immunochemotherapeutic that has potential utility for improved therapeutic resolution of this neoplastic condition. Potential fludarabine molecular delivery platforms relevant to B-CLL include anti-CCR7, anti-CXCR5, CD120a (TNFR1), anti-CD19, anti-CD20 and anti-CD52. One advantage of considering covalent immunochemotherapeutics like fludarabine-(C2-methylhydroxyphosphoramide)- [anti-IGF-1R] for leukemia conditions is that at least theoretically, a relatively large percentage of the total dose (chemotherapeutic moiety) has access to and rapidly comes in direct physical contact with a greater percentage of metastatically transformed neoplastic cells compared to “solid tumor” neoplastic disease states. In turn, fludarabine-( C2-methylhydroxyphosphoramide)-[anti-IGF-1R] or other covalent immuno chemotherapeutics bound to the external surface membrane of leukemia cell populations may more effectively initiate and be more susceptible to cytotoxic resolution by host immune responses associated with cytoxic mechanisms of ADCC, complement mediated cytolysis and opsonization/phagocytosis. Each of these advantages and attributes is complemented by the potential for fludarabine-( C2-methylhydroxyphosphoramide)-[anti-IGF-1R] to produce higher cytosol fludarabine concentrations than are possible following simple intravenous injection; a prolongation of the plasma fludarabine pharmacokinetic profile; and the opportunity to evoke synergistic or additive levels of selective anti-neoplastic cytotoxicity through simultaneous dual mechanisms-of-action (e.g. fludarabine in combination with anti-trophic receptor monoclonal immunoglobulin). | |
| Implementation of immunoglobulin as a molecular platform for covalent immunochemotherapeutics imparts several other properties that significantly complement the selective binding avidity of IgG which facilitates the characteristics of selective “targeted” chemotherapeutic delivery. Many of these attributes are directly associated with the physical size of the immunoglobulin molecule (150-kDa). Although the properties of selective “targeted” delivery properties for the covalent immunochemotherapeutic, fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] contribute to significantly large declines in innocent exposure of normal tissues and healthy organ systems to the fludarabine moiety, it is the relatively massive size the immunoglobulin component that is probably most responsible for facilitates this attribute. More specifically, the immunoglobulin component of fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] substantially reduces or prevents passive diffusion of the fludarabine moiety across intact cellular external membrane structures. The combined features of selective “targeted” delivery and relatively large molecular weight can collectively contribute to providing a margin-of-safety for fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] that allow it be be administered for a longer period of time and potentially at higher fludarabine-equivalent dosases compared to fludrabine chemotherapeutic. Presumably, at least to some degree, steric hinderance phenomenon associated with the relatively large size of the immunoglobulin molecule is responsible for covalently bound chemotherapeutics being less vulnerable to the function of P-glycoprotein [110-113] which is the non-selective trans-membrane efflux “pump” (MDR-1: multi-drug resistance protein) [16] most commonly responsible for chemotherapeutic-resistance [113-118]. The relatively large molecular weight of the anti-IGF-1R component of fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R] also reduces the rate and extent that fludarabine enters the renal plasma filtrate and is subsequently excreted into the urine (glomerular MWCO=60-kDa, IgG=150-kDa). By delaying the rate and extent of excretion by renal mechanisms, then technically the fludarabine pharmacokinetic profile becomes significantly prolonged or extended which further facilitates continual selective membrane deposition and intra-cellular accumulation of the fludarabine within “targeted” neoplastic cell populations. | |
Conclusion |
|
| The molecular design and the corresponding organic chemistry reactions for covalently bonding fludarabine to immunoglobulin or other biologically relevant protein fractions has not previously been described in published reports. Attributes of the organic chemistry reactions scheme for the synthesis of the Phase-III fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] end-product include; [i] a relatively brief reaction time for synthesizing the Phase-III endproduct from the Phase-II amine reactive intermediate; [ii] option of generating a stable Phase-II fludarabine amine-reactive intermediate if an anhydrous Phase I & II solvent system is applied (e.g. DMSO, DMF); [iii] flexibility of utilizing other phosphate chemotherapeutic analogs; and [iv] ability to substitute other biologically relevant protein fractions in place of anti-IGF-1R; and comparatively low level of dependency on advanced forms of instrumentation. Physical and functional attributes of the final Phase-III fludarabine-(C2- methylhydroxyphosphoramide)-[anti-IGF-1R] end-product are; [i] molar-incorporation-index of 3.67:1; [ii] lack of any “foreign” or artificial chemical groups introduced into the final Phase-III end-product; [iii] retained IGF-1R binding-avidity; [iv] absence of any detectable IgG-IgG polymerization or low molecular weight fragmentation. The lack of any 5-carbon or 6-carbon ring structures inserted into fludarabine-(C2-methylhydroxyphosphoramide)-[anti- IGF-1R] end-product during the course of implementing the organic chemistry reactions in the multi-phase synthesis regimen reduces the probability of inducing subsequent host humoral immune responses. | |
| Covalent fludarabine immuno chemotherapeutics potentially can provide a spectrum of desirable attributes that are not possible with conventional small molecular weight chemotherapeutic agents. Most important in this regard is their ability to promote or facilitate; [i] selective and continual chemotherapeutic deposition on the exterior surface membrane of neoplastic cells; [ii] progressive chemotherapeutic accumulation within the cytosol/intracellular compartment of neoplastic cells to concentrations that are 8.5X to 100X greater than can be attained by simple passive diffusion; [iii] reducing the effectiveness of chemotherapeutic resistance mechanisms (e.g. P-glycoprotein); [iv] potential opportunity to attain synergistic or additive anti-neoplastic cytotoxicity in a single biopharmaceutical agent; [v] reduced innocent chemotherapeutic exposure of normal tissues and healthy organ systems; [vi] prolongation of plasma chemotherapeutic pharmacokinetic profiles; and [vii] reduced burden on metabolization pathways and excretion processes. | |
References |
|
|
|