Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Forensic Toxicol Pharmacol Vol: 2 Issue: 2
Screening of Urine and Blood Using Limited Sample Preparation and Information Dependent Acquisition with LC-MS/MS as Alternative for Immunoassays in Forensic Toxicology
| Ruth Verplaetse*, Sylvie Decabooter, Eva Cuypers and Jan Tytgat | |
| University of Leuven, KU Leuven, Toxicology and Pharmacology, Campus Gasthuisberg O&N 2, Herestraat 49, 3000 Leuven, Belgium | |
| Corresponding author : Dr. Ruth Verplaetse University of Leuven, KU Leuven, Toxicology and Pharmacology, Campus Gasthuisberg O&N 2, PO Box 922, Herestraat 49, 3000 Leuven, Belgium Tel: +32 16 323411; Fax: +32 16 323405 E-mail: ruth.verplaetse@pharm.kuleuven.be |
|
| Received: August 22, 2013 Accepted: October 22, 2013 Published: October 26, 2013 | |
| Citation: Verplaetse R, Decabooter S, Cuypers E, Tytgat J (2013) Screening of Urine and Blood Using Limited Sample Preparation and Information Dependent Acquisition with LC-MS/MS as Alternative for Immunoassays in Forensic Toxicology. J Forensic Toxicol Pharmacol 2:2. doi:10.4172/2325-9841.1000112 |
Abstract
Screening of Urine and Blood Using Limited Sample Preparation and Information Dependent Acquisition with LC-MS/MS as Alternative for Immunoassays in Forensic Toxicology
Immunoassays are widely used to perform an initial toxicological screening of biological samples. However, LC-MS/MS is described as a promising technique that can overcome the limitations of immunoassays (such as their lack of selectivity). The objective of this project was to implement a LC-MS/MS method for screening of forensic ante- and post-mortem urine and whole blood samples that can replace the immunoassays. Easy and rapid sample preparation techniqueswere evaluated. Protein precipitation with acetonitrile combined with aqueous dilution (dilution factor 5 for urine and 10 for blood) proved to be an effective procedure. On the LC-MS/MS, 1 scheduled multiple reaction monitoring transition for each of 414 compounds was analyzed in positive mode, followed by an enhanced product ion scan if the peak height exceeded a specified threshold.
Keywords: Forensic; Toxicology; Screening; Urine; Whole blood; LC-MS/MS; Immunoassay
Keywords |
|
| Forensic; Toxicology; Screening; Urine; Whole blood; LC-MS/MS; Immunoassay | |
Abbreviations |
|
| CE: Collision Energy; DAD: Diode Array Detection; EPI: Enhanced Product Ion Scan; GC: Gas Chromatography; LC: Liquid Chromatography; LLE: Liquid-Liquid Extraction; IDA: Information Dependent Acquisition; IS: Internal Standard; ME%: Matrix Effects; MRM: Multiple Reaction Monitoring; (MS/)MS: (tandem) Mass Spectrometry; RE%: Recovery; RT: Retention Time; sMRM: scheduled Multiple Reaction Monitoring; SPE: Solid-Phase Extraction; PE%: Process Efficiency | |
Introduction |
|
| Immunoassays are widely used for screening of biological samples. In case of a positive result, an additional selective confirmation analysis is performed. Immunoassays are simple and quick, but costly. Moreover, immunoassays are not selective: no individualistic compound (e.g. diazepam), but only a group (e.g. benzodiazepines) is detected. Besides the compounds included in a group, other structurally related compounds can result in a false positive test because of cross-reactivity. Not all drug classes are covered by the immunoassays and some systems will disappear from the market (e.g. Abott Axsym® which is routinely used for screening of forensic (i.e. both ante- and post-mortem) urine samples in our laboratory). Therefore, we searched for an alternative for the immunoassays for screening of forensic urine and whole blood samples. | |
| Two promising approaches using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) for screening are described: multi-target screening and general unknown screening [1-6]. In the first approach only a selected group of compounds is detected, while this is not limited in the second approach. As a consequence, sensitivity of the multi-target screening is in general higher, but the number of compounds that can be detected is lower. For multi-target screening, a triple quadrupole, ion trap or a hybrid instrument combining these two are preferred as the mass analyzer. For general unknown screening, measuring the accurate mass of compounds using a time-of-flight mass analyzeris becoming more and more popular. | |
| Several simple sample preparation methods for screening with LCMS/ MS are described [1-6]. For urine, dilution with different dilution factors and solvents, protein precipitation with several precipitating agents and filtration are used as simple alternatives for more complex liquid-liquid extraction (LLE) and solid-phase extraction (SPE) [1-5]. Screening of whole blood (especially post-mortem) is more difficult than urine or serum/plasma because of the complexity of this matrix. This explains why almost all LC-MS/MS methods for screening of whole blood use SPE or LLE as sample preparation [1-3]. There is only one paper describing a protein precipitation procedure (including an evaporation step) for screening of post-mortem whole blood [6]. Clearly, research on easy and quick sample preparation for whole blood is very limited. Moreover, to the best of our knowledge, there is no publication that describes LC-MS/MS screening of both ante- and post-mortem urine and whole blood. | |
| The objective of this project was to develop an easy, quick and low-cost sample preparation and LC-MS/MS method for screening of forensic urine and whole blood samples. | |
Materials and Methods |
|
| Chemicals, standards and samples | |
| CON-DOAm®containing known concentrations of amphetamine, benzoylecgonine, codeine, dextropropoxyphene, methamphetamine, methadone, methaqualone, morphine, oxazepam and phencyclidine was purchased from Siemens Medical Solutions Diagnostics (LA, USA). The internal standard (IS) N-methylclonazepam was purchased from LGC (Molsheim, France). Acetone was purchased from Merck (Darmstadt, Germany). Water was obtained from a Milli Q Water Purification System (Millipore, Brussel, Belgium). LCMS grade acetonitrile was purchased from Biosolve (Valkenswaard, The Netherlands). All LC-MS grade mobile phase additives (formic acid and ammonium formate) were purchased from Sigma- Aldrich (Bornem, Belgium). Glassware was silanized using AquaSil Siliconizing Fluid (Thermo Scientific, Breda, The Netherlands). 1.5 mL screw cap vials, 100 μL inactivated glass vial inserts, Whatman® Mini- UniPrep™ syringeless filters with 0.2 μm or 0.45 μm PTFE filtration membranes and Toxitubes® were purchased from Agilent (Diegem, Belgium). Statistical analyses were performed with GraphPad Prism (version 6.01, La Jolla, US). | |
| Immunoassays | |
| For screening of urine samples, a fluorescence polarization immunoassay (Abbott Axsym® system, Waver, Belgium) was used to detect amphetamine/metamphetamine, barbiturates, benzodiazepines, cannabinoids, cocaine and metabolites, methadone, opiates and tricyclic antidepressants. Used cutoffs were 50 ng/ mL for cannabinoids and tricyclic antidepressants, 60 ng/mL for benzodiazepines, 100 ng/mL for methadone, 150 ng/mL for cocaine, 200 ng/mL for barbiturates and opiates, 500 ng/mL for (met) amphetamine. Screening of blood samples was performed with an enzyme immunoassay (Cozart® system, Oxfordshire, UK) which detected the presence of amphetamine/metamphetamine, barbiturates, benzodiazepines, cannabinoids, cocaine and metabolites, methadone and opiates. Used cutoffs were 5 ng/mL for methadone, 10 ng/mL for cannabinoids, 20 ng/mL for barbiturates, 50 ng/mL for cocaine and metabolites, 100 ng/mL for (met)amphetamine, benzodiazepines and opiates. | |
| Sample preparation for LC-MS/MS screening | |
| Seven sample preparations were tested for screening of urine and blood: (1,2) precipitation with acetonitrile/acetone and aqueous dilution; (3,4,5,6) protein precipitation with acetonitrile/acetone, aqueous dilution and 0.2/0.45 μm filtration and (7) a simple LLE. For the precipitation procedures, 200 μL precipitating solvent (acetonitrile or acetone containing 500 ng/mL N-methylclonazepam) was slowly dropped to 100 μL supernatant of urine (150 μL urine was centrifuged for 10 min, 1000 × g) or blood while vortexing. After centrifugation (10 min, 1000 × g) the supernatant was diluted with 700 or 300 μL water (equal to a dilution factor 10 or 5) in a 1.5 mL vial or in the chamber of a Whatman® Mini-UniPrep™ syringeless filter (in this case volumes were decreased by half as the maximum volume of the filter is 500 μL). The plunger containing the 0.2 μm or 0.45 μm PTFE filtration membrane was manually pressed through the diluted sample into the chamber and the filtrate is forced into the reservoir of the plunger. The Mini-UniPrep filter was then placed in the autosampler of the LCMS/ MS. Compared to the common use of a syringe and filters, this syringeless system reduces waste and avoids contamination. For the LLE, the Toxitubes (Toxitubes A for extraction of basic and neutral drugs and Toxitubes B for extraction of acidic and neutral drugs) were vortexed for 10 s. Next, 100 μL urine or blood, 200 μL IS (500 ng/mL N-methylclonazepam in H2O) and 4.7 mL water (in Toxitube A) or 4.2 mL water (in Toxitube B) were added. The tubes were mixed for 5 min and centrifuged (5 min, 1000 × g). The organic layer was evaporated to dryness at room temperature. The sample was reconstituted with 200 μL acetonitrile and 800 μL water and transferred into a 1.5 mL vial (equal to a dilution factor 10). | |
| LC-MS/MS | |
| LC-MS/MS analysis was carried out using an UFLC Shimadzu system consisting of 2 LC-20ADXR pumps, a SIL-20ACXR autosampler, a DGU-20A3 degasser and a CTO-20A oven (Shimadzu Prominence, Antwerpen, Belgium) in combination with a 3200 QTRAP (ABSciex, Halle, Belgium) and Analyst software (version 1.5). An existing multi-target screening approach for this kind of apparatus was adapted from the literature: a scheduled multiple reaction monitoring-information dependent acquisition-enhanced product ion (sMRM-IDA-EPI) multi-target screening approach [4]. The method starts with a survey sMRM scan, where MRM transitions are only monitored during the expected retention time window. When a sMRM signal exceeds a preset IDA-threshold, an EPI scan is performed (this is a product scan where the third quadrupole is used as a linear ion trap). However, the original sMRM-IDA-EPI screening method is only performed in positive ionization and situations occur where compounds cannot be identified because the MRM signal is too low to trigger an EPI scan or because the EPI quality is insufficient for identification [4]. The existing method was optimized to overcome these shortcomings. The number of detected compounds was reduced from 700 to only 414 forensic relevant compounds (Table 1). An extra sMRM method in negative ionization mode measuring two MRM transitions for 38 compounds was added in order to detect a broader range of compounds (Table 2). However, the positive and negative method were not sensitive enough for detection of cannabis use, as shown by preliminary comparison of LC-MS/MS and immunoassays for some real-life forensic samples of cannabis users. To enhance the sensitivity of the screening for detection of cannabis, an important drug in forensic investigations, a MRM method for analysis of THC, THC-OH and THC-COOH was added (Table 2). In summary, each sample was injected three times and analyzed by three LC-MS/MS methods: (1) a sMRM-IDA-EPI method in positive ionization mode, (2) a sMRM method in negative mode and (3) a positive MRM method for detection of THC, THC-OH and THC-COOH. | |
| Table 1: Analytes measured in the positive sMRM-IDA-EPI method. | |
| Table 2: Analytes, their retention time (RT) and MRM parameters (Q1, Q2 and collision energy (CE)) measured in the negative sMRM method for detection of 38 compounds and the positive MRM method for detection of THC and metabolites. | |
| LC conditions | |
| The used pentafluorophenyl propyl column (5.0 μm particle size, 2.1 mm x 50 mm), fitted with a guard column (2.1 mm × 10 mm, same packing material) and a filter of 2.0 μm, was purchased from Restek (via Interscience, Louvain-la-Neuve, Belgium). The autosampler temperature was set at 15°C, the column oven at 40°C. The autosampler needle was rinsed before and after sample injection to avoid carry over. The mobile phase consisted of water with 2 mM ammonium formate and 0.2% formic acid (A) and acetonitrile with 2 mM ammonium formate and 0.2% formic acid (B). The positive sMRM-IDA-EPI and negative sMRM method had following gradient conditions: 0-10 min: 10-90%B and increase of flow rate from 0.5 mL/ min to 1 mL/min; 10-15 min: 90%B at 1 mL/min; 15-15.5 min: 90- 10%B; 15.5-17.5 min: 10%B at 0.5 mL/min. Following gradient at a flow rate of 0.5 mL/min was used in the MRM method: 0-10 min: 10-90%B; 10-11 min: 90%B; 11-11.5 min: 90-10%B; 11.5-13.5 min: 10%B. The injection volume was 30 μL. | |
| MS/MS conditions | |
| For all three LC-MS/MS experiments, electrospray conditions were as follows: gas 1: nitrogen, 40 psi; gas 2: nitrogen, 70 psi; ion-spray voltage: 4000 V (-4000 V in negative mode); ion-source temperature: 500°C; curtain gas: nitrogen, 20 psi; collision gas: high. The declustering potential was 40 V (-40 V in negative mode), the entrance potential 10 V (-10 V in negative mode), the cell exit potential 5 V (-5 V in negative mode). Q1 and Q3 were operated in unit resolution. In the sMRM-IDA-API method, 414 MRM are measured ±90 s around the expected retention time of the compound (Table 1). The target scan time was 1 s with a pause between the sMRM transitions of 2 ms. The used sMRM transitions were adapted from literature [4]. If the sMRM peak height exceeded the IDA-treshold (1000 counts per second (cps)) an EPI scan was triggered for the two most abundant sMRM signals. sMRM transitions which triggered the EPI scan twice consecutively were excluded for EPI scans for 15 s. The EPI scans were performed in a mass range from 50 to 640 Da at 4000 Da/s applying a collision energy (CE) of 35 ± 15 V, a fixed fill time of 50 ms and Q0 trapping. The acquired EPI spectra were automatically compared to the used ABSciex MS/MS library containing 1253 compounds [7]. In negative ionization mode, a sMRM method was used containing 2 sMRM transitions for each of 38 compounds (Table 2). Each MRM is measured ± 90 s around the expected retention time of the compound. For analysis of THC, THC-OH and THC-COOH a separate MRM method with dwell times of 30 ms was used (Table 2). | |
| LC-MS/MS data analysis | |
| For the sMRM-IDA-EPI method, a report was automatically generated. However, for accurate identification, manual review of the data is necessary [5]. Following criteria were used for identification: (1) the blank must be negative, (2) the IS must be present at the correct retention time with purity >75% and (3) the 4 (3 if ≤5 ions in the library spectrum) most abundant ions in the library spectrum must be present in the unknown spectrum. Ions present in the unknown spectrum that are not present in the library spectrum must be smaller than the 2 (3 if ≤5 ions in the library spectrum) most abundant ions in the unknown spectrum. For the (s)MRM methods, retention time, presence of the MRM transitions and ratio between MRM transitions were used for identification. | |
| Method validation | |
| The process efficiency (PE%) that includes the influence of possible matrix effects (ME%) and recovery (RE%) was calculated. Two sets of samples were prepared for determination of PE%. In set 1, blank matrices (5 different sources of both urine and whole blood) were spiked with pure standard before sample preparation. Set 2 consisted of pure standards. Fifteen compounds with varying characteristics representing the broad range of compounds detected in this screening were carefully selected. Following formula was used to calculate the PE%: | |
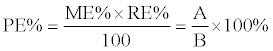 |
|
| Where A is de peak area of the measured MRM transition from set 1, B from set 2. A value of 100% reflects the perfect situation. Clear guidelines on the acceptability of PE% values do not exist, so we had a look at the acceptance criteria for ME% and RE%. Acceptance criteria for ME% are set to 75-125% with a coefficient of variation (CV%= standard deviation divided by average) of maximum 15% (or 20% near the limit of detection) [8]. RE% is acceptable if the CV% is smaller than 15% (or 20% near the limit of detection) [8]. However, for qualitative methods acceptance criteria could be less strict. Selectivity was evaluated with blank samples from different sources (n=10 each for both urine and blood, no IS was added during sample preparation) and zero samples (n=2, IS was added to blank samples during sample preparation). To determine carryover, 30 μL H2O was injected as blank after every sample. Finally, 162 forensic urine and 146 whole blood samples were analyzed using the LC-MS/ MS method and compared with results from immunoassays and confirmation analyses, describing the accuracy, selectivity, sensitivity and carryover of the sample preparation and LC-MS/MS method. With each batch, a quality control sample (blood or urine spiked with CON-DOA) was run to check overall system performance. | |
Results and Discussion |
|
| Seven sample preparations (resulting in a 10-fold dilution of the urine and blood samples) were tested for ten compounds with varying characteristics representing the broad spectrum of compounds that can be found in forensic samples (amphetamine, benzoylecgonine, codeine, dextropropoxyphene, methamphetamine, methadone, methaqualone, morphine, oxazepam and phencyclidine) (Figure 1). Precipitation, aqueous dilution and filtration had an equal or lower PE% than the same procedure without filtration (Figure 1(2 versus 1)). The LLE using the Toxitubes was highly variable and showed a low PE% for most of the compounds (Figure 1(3)). Samples treated with acetonitrile had in general less variability than samples treated with acetone (Figure 1(A versus B)). There was no significant difference between filtration membranes of 0.45 μm and 0.2 μm (Figure 1(* versus °)). Based on the following three criteria (i.e. PE% around 100%, low variability and ease of use), protein precipitation with acetonitrile and aqueous dilution was selected as the optimal sample preparation (Figure 1(1A)). | |
| Figure 1: PE% of the different sample preparations tested for ten compounds in urine (U) and whole blood (B). Tested methods included: precipitation with acetonitrile and 10-fold dilution (1A), precipitation with acetone and 10-fold dilution (1B), protein precipitation with acetonitrile, 10-fold dilution and 0.2 µm filtration (2A°), protein precipitation with acetonitrile, 10-fold dilution and 0.45 µm filtration (2A*), protein precipitation with acetone, 10-fold dilution and 0.2 µm filtration (2B°), protein precipitation with acetone, 10-fold dilution and 0.45 µm filtration (2B*) and LLE using the Toxitubes (equal to a dilution factor 10) (3). The average and standard deviation of 10 measurements (5 different sources of both urine and whole blood, analyzed in duplicate) are shown. The numbers of the sample preparations that are significantly different are noted above each sample preparation (one-way ANOVA, Tukey-Kramer test, p<0.05). Tested concentrations in urine and whole blood were 2.5 µg/mL amphetamine, 0.75 µg/mL benzoylecgonine, 1.5 µg/ mL codeine, 0.375 µg/mL dextropropoxyphene, 2.5 µg/mL methamphetamine, 0.5 µg/mL methadone, 0.9 µg/mL methaqualone, 0.15 µg/mL morphine, 0.5 µg/mL oxazepam and 0.125 µg/mL phencyclidine. | |
| Since urine samples are in general cleaner and have a lower viscosity than whole blood samples, we tried to lower the dilution factor in order to enhance sensitivity. We tested undiluted urine, dilution factor 5 and 10 for the screening of spiked urine samples (see values for “urine, high”, Table 3). As expected, significant and highly variable matrix effects were seen when directly injecting urine after centrifugation. Moreover, a shift in retention time from run to run was seen as a consequence of contamination of the LC column. Fiveand 10-fold dilution had better PE% values and lower variability. As a compromise between sensitivity and repeatability, protein precipitation with acetonitrile and 5-fold dilution was selected as the sample preparation for urine. | |
| Table 3: Fraction bound to plasma protein (Fb), PE and CV for 15 compounds in urine and blood analyzed with the LC-MS/MS screening. | |
| The PE% of the selected sample preparations (i.e. protein precipitation with acetonitrile and 5-fold dilution for urine and protein precipitation with acetonitrile and 10-fold dilution for blood) was further measured in both matrices for the ten compounds at different concentration levels, to ensure its analytical quality at different concentrations and in blood for five substances almost 100% bound to plasma proteins (see values in italic, Table 3). These five (buprenorphine, clomipramine, cocaine, midazolam and zolpidem) were included to ensure that the used sample preparation works for all compounds. The lowest PE% can be expected in blood, where the protein precipitation is more drastic than in urine. Drugs that are highly bound to proteins in the blood have an increased risk of being lost in the precipitant. Therefore, their behavior was tested. All PE% values were higher than 60% with CV% smaller than 30%, except for morphine in urine (see values in italic, Table 3). The obtained PE% values were considered acceptable for screening. The method was found to be selective. No carryover was seen. | |
| Finally, the optimized sample preparation and three LC-MS/MS methodswere used for screening of 162 forensic urine samples and 146 whole blood samples (both ante- and post-mortem species). These samples were also analyzed with the appropriate immunoassay and confirmation techniques routinely used in the laboratory (Table 4). To evaluate LC-MS/MS as an alternative tool to immunoassay screening, the number of true and false positive and negative results obtained by both techniques was compared. For urine samples, screening by immunoassay resulted in a higher number of false positives (44 versus 2) and false negatives (55 versus 21) than screening by LCMS/ MS (Table 4). For blood samples, the immunoassay also had a higher number of false negatives (67 versus 23) and false positives (4 versus 1) (Table 4). The Cozart® immunoassay had a lower number of false positives than the Axsym® system, as the Cozart® tests include less compounds in each class. The higher number of false positives for urine samples analyzed by the immunoassay can be explained by the low selectivity of the immunoassay: there is crossreactivity with molecules structurally related to the target analyte(s) (e.g. in post-mortem samples, amphetamine-like compounds can be present because of putrefaction, generating a false positive result in the immunoassay). The higher number of false positives results in a higher cost (since more confirmation tests are required). False negative results were seen for samples containing low levels of drugs or if a compound is just not detected by the immunoassay (e.g. the Cozart® test for opiates does not react with fentanyl or tramadol which are detected by LC-MS/MS). Evidently, false negatives should be avoided in forensic toxicology. Considering the number of true and false results, LC-MS/MS screening was more specific, sensitive and correct than the immunoassay. The saving of time of screening by LC-MS/MS compared to the immunoassays was limited because of the need to analyze each sample with three different LC-MS/MS methods. However, because of the gain in efficiency, analysis time was considered of secondary importance. | |
| Table 4: Real-life forensic urine and blood samples screened with immunoassays and LC-MS/MS. | |
Conclusion |
|
| In the search for an alternative for screening by immunoassays, several sample preparations of urine and whole blood followed by LC-MS/MS analysis were evaluated. Protein precipitation using acetonitrile combined with aqueous dilution (dilution factor 5 for urine and 10 for blood) proved to be an effective sample preparation for screening using LC-MS/MS. The samples were analyzed by three LC-MS/MS methods (sMRM-IDA-EPI for 414 positively charged compounds, negative sMRM for 38 acidic compounds and positive MRM method for THC and two metabolites) in order to cover a broad range of forensic relevant compounds. To the best of our knowledge this is the first LC-MS/MS method for screening of both ante- and post-mortem urine and whole blood. For 162 forensic urine samples and 146 whole blood samples, screening by LC-MS/MS performed superiorly compared to the immunoassays. Hence, LC-MS/MS can be considered a trustworthy alternative to immunoassays for screening in forensic toxicology. | |
Acknowledgments |
|
| The authors are grateful to Sara Loix, Arne Bonneure and Dieter Waumans for their help. | |
References |
|
|
|