Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Short Communication, J Nanomater Mol Nanotechnol Vol: 5 Issue: 2
An Alternative Way to Prepare Biocompatible Nanotags with Increased Reproducibility of Results
| Mohammad Salehi1*, Wilrun Mittelstädt2, Jens Packeisen3,Markus Haase2 and Angela Hamann-Steinmeier1 | |
| 1Faculty of Engineering and Computer Science, University of Applied Science Osnabrueck, Artilleriestr. 46 D- 49076 Osnabruck, Germany | |
| 2Facility of Biology and Chemistry, Barbarastr. 7, D-49069 Osnabruck, Germany | |
| 3Medical Center, Rostocker Strasse 5-7, D-49124 Georgsmarienhütte, Germany | |
| Corresponding author : Mohammad Salehi Faculty of Engineering and Computer Science, University of Applied Science Osnabrueck, Artilleriestr. 46, D- 49076 Osnabrueck, Germany E-mail: msalehi@uos.de |
|
| Received: January 25, 2016 Accepted: April 14, 2016 Published: April 20,2016 | |
| Citation: Salehi M, Mittelstädt W, Packeisen J, Haase M, Hamann-Steinmeier A (2016) An Alternative Way to Prepare Biocompatible Nanotags with Increased Reproducibility of Results. J Nanomater Mol Nanotechnol 5:2. doi:10.4172/2324-8777.1000181 |
Abstract
An Alternative Way to Prepare Biocompatible Nanotags with Increased Reproducibility of Results
Aggregation of nanoparticles (NP) occurs mostly during and after their biofunctionalization. Aggregations can lead to disfunction of the biomolecule which binds to NP like antibodies or enzymes. In many cases, disfunctions and the lack of reproducibility of the results are two major challenges during the use of nanotechnology. We demonstrate that the aggregation of nanoparticles can be controlled by choosing the optimal amount of crosslinker and furthermore the function of biomolecules can be protected by avoiding centrifugation steps. These investigations may provide a valuable guide in many other medical applications of nanostructures.
Keywords: Raman; SERS; Preparation of metallic nanoparticles; Imaging; Immunodetection; Nanotags; Biocompatibility of Nanotags; Immunohistochemistry; SERS-microscopy
Keywords |
|
| Raman; SERS; Preparation of metallic nanoparticles; Imaging; Immunodetection; Nanotags; Biocompatibility of Nanotags; Immunohistochemistry; SERS-microscopy | |
Introduction |
|
| Currently, nanotechnology is successfully used in numerous different medical applications [1]. Especially NPs with diameters between 1-100 nm are ideal tools for diagnostic applications in medicine because of their large surface to volume ratio [2]. Surface Enhanced Raman Scattering (SERS) is an ultrasensitive method for the investigation of molecules in close proximity to plasmonic metallic nanostructures and plays an important role in nanomedicine [1,3]. In comparison to other established labeling approaches such as fluorophores, dyes or enzymes, the SERS methodology offers several unique advantages in analytical and life sciences. One key benefit of SERS labels is their spectral multiplexing capacity due to the small line width of vibrational Raman bands. Furthermore, only a single laser line is needed for simultaneously exciting the spectrum of several SERS labels [1,4]. The major disadvantage of SERS methods, similar to other nanotechnology based methods could be the low reproducibility of results [5]. Previous studies report instability of NPs reacting with salt especially during transfer of NPs in biological buffers before and during their application [6]. On the other hand, proteins such as immunoglobulins (IgGs) have the same attribute to form aggregations just as NPs [7]. The pivotal factor for success of biomedical tasks depends on the quality of each involved element like IgGs and nanostructures before, during and after their conjunction. Reproducibility of results depends directly on the stability of nanoproducts after their preparation or during the measurements. Especially the binding affinity of IgG coupled NPs depends on the degree of NP aggregation. Hence we call the NPs that maintain their biofunction “biocompatible NPs”. | |
| biofunction “biocompatible NPs”. The loss of target accuracy and appearance of nonspecific binding are most important disadvantages of non-magnetic nanoparticles. The detection methods that are based on magnetic microparticles show better reproducibility, although the micropaticles are heavier than 1-100 nm gold nanoparticles (GNP). The major difference between these methodologies is centrifugation steps to remove an excess of chemicals or unbound proteins such as IgGs. During the preparation of spherical monodisperse nanoparticles a size deviation around 5% is acceptable [8]. However, this small difference in size has a huge effect on the weight of the produced nanoparticles. For example the weight distribution of produced 55 nm GNP is around 30% (r=27.5 ± 1.375 nm). Their weight is as for the weight of a sphere in direct correlation to its volume | |
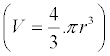 |
|
Materials and Methods |
|
| Silver nitrate (AgNO3), polyvinylpyrrolidone K10 and K30 (PVP), tetrachloroauric (III) acid (HAuCl4), 5,5’-dithiobis (2-nitrobenzoic acid) (DTNB), sodium borohydride, bovine serum albumin (BSA), N-(3-dimethylaminopropyl)-N’-ethyl carbodiimide (EDC), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), Hepes (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), acetic acid, Tris/HCl, KCl, NaCl, Na2HPO4, KH2PO4, glycine, Ponceau S, Mercaptohexadecanoic acid, Methanol and Ethanol were purchased from Sigma/Aldrich/Fluka. Monoclonal mouse antibody p63 was purchased from Santa Cruz Biotechnology. Polyclonal HRP-IgG was purchased from Abcam (Germany). Peroxidase-conjugated goat α-mouse antibody was purchased from Jackson ImmunoResearch. Standard transfer buffer for immunoblotting (25 mM tris, 192 mM glycine, 10% methanol, pH 8.3) and Ponceau S staining solution (0.2% Ponceau S, 1% acetic acid) were prepared. Pierce ECL western blotting substrate was purchased from Thermo Scientific. Phosphatebuffered saline (PBS) and Tris-buffered saline (TBS) were prepared according to standard protocols, using 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 (pH=7.4), and 20 mM tris/HCl (pH=7.6), 685 mM NaCl, respectively. All chemical reagents were used without further purification. Ultrapure water (18 MΩ) was used in all reaction steps. | |
| Preparation of antibody-SERS label conjugates | |
| Gold nanoparticles: Gold nanoparticles (GNP) with an average size of 55 nm and 60 nm (± 2.5 nm) were synthesized by procedures reported previously [9]. The preparation of Au@Ag is based on the template-engaged replacement reaction between silver nanoparticles and HAuCl4 according to the method described by Sun and coworkers [10,11]. | |
| Synthesis of SERS labels: 2-nitro-5-thiobenzoate (4-NTB) was obtained by cleavage of DTNB with sodium borohydride and was used as Raman label. 16-MHA (16-Mercaptohexadecanoic acid) with terminal reactive group (COOH) was used for bioconjugation. Surface functionalization of nanoparticles (Au@Ag or GNP) was achieved by incubation with a 99:1 mixture of 4-NTB and 16-MHA, which yields a complete self-assembled monolayer (SAM) on the particle surface (Figure S1) [4,12]. The reaction of the SERS labels with EDC and sulfo-NHS yields the corresponding NHS ester, i.e. an activated SERS label. The optimal amount of EDC/sulfo-NHS was determined using the dilution of stock solution with known concentrations of each one. The stock solutions of 1.6% s-NHS and 2,4% EDC were prepared in a 50 mM Hepes buffer and serially diluted (four times, 1:2 ) in 50 mM Hepes. 50 μl of each s-NHS and 50 μl of EDC solutions were added to 500 μl SERS labels (OD=1) in 50 mM Hepes buffer. The NPs were incubated at room temperature (RT) and gently vortexed for around 20-25 minutes. Afterwards the NPs were centrifuged (5000 g, 10 minutes) and suspended in 500 μL of Hepes buffer. One μg HRP-IgG was added to the activated GNP labels for an incubation time of 60 min at room temperature. The colloid was washed two times with a mixture of PBS and 0.2% BSA. The extinction spectrum of samples was measured after the last conjugation and is shown in Figure S2. Au@Ag was used as SERS labels for imaging the tumor suppressor p63 in prostate biopsies. 50 μl 0.2% s-NHS solution and 50 μl of a 0.3% EDC solution were added to 500 μl SERS labels in 50 mM Hepes buffer [12-14]. Vacuum (3 minutes, range of -500 to -600 mbar) system with two chambers was used to remove the exess of crosslinker or IgG (pore size of membrane=150 kDa) [15]. One μg anti p63 IgG was added to activated SERS NP after removing the crosslinker and was incubated for 60 min at room temperature. Then to remove the excess of unbounded IgG, under vacuum (3 minutes, range of - 500 to - 600 mbar), 4.5 ml Hepes was passed through the membrane (pore size of membrane=150 kDa). Finally, the SERS labeled IgG were dispersed in PBS buffer with 0.2% BSA [13,14]. | |
| Ponceau staining and western blot: SDS-PAGE was performed according to standard protocols. Semi-dry blotting was carried out at 10 V constant for 45 minutes. The membrane was incubated with Ponceau S staining solution for three minutes and rinsed with ultrapure water for contrast. After this non-specific sites were blocked with 5% milk powder in TBS-T (0.1% Tween 20 in TBS) for 30 minutes at room temperature. The membrane was briefly rinsed with ultrapure water and incubated with secondary antibody (peroxidase-conjugated goat α-mouse; diluted 1:10,000 in 5% BSA in TBS-T) for 1 hour. Following three washing steps (15 minutes each) with TBS-T the signals were detected using ECL western blotting substrate according to the manufacturer’s protocol and read out at the ChemiDoc MP System (Bio-Rad). | |
| Tissue preparation and incubation with SERS labels: Formalinfixed and paraffin-embedded prostate tissue specimens from patients undergoing prostatectomy were mounted as 4 μm thick sections on glass slides. After paraffin removal and antigen retrieval (target retrieval solution from Dako), the tissue sections were incubated for 10 minutes with blocking solution (1% BSA in PBS). The tissue sections were incubated with 200 μl SERS labeled p63 antibody (OD=0.2) in a PBS buffer with 0.1% BSA for 20 minutes. Unbound NP-labeled IgGs were removed by several washing steps (PBS buffer with 0.1% BSA) [13,14]. | |
| Instruments | |
| Raman microspectroscopy: Raman experiments were performed with a Raman microspectrometer (WITec, Alpha 300R) comprising a microscope, a monochromator with a 600-grooves/mm grating, and a Peltier-cooled CCD camera. Radiation from the 632.8-nm line of a HeNe laser was focused on the sample by a 40× microscope objective. SERS mapping experiment were performed with 0.15 s integration time per pixel, a step size of 1 μm and 6 mW laser at the sample. | |
| Extinction spectroscopy: Extinction spectra were measured in a 10-mm quartz cuvette with a Lambda 35 UV/VIS absorption spectrometer (PerkinElmer). | |
| Transmission electron microscopy: A transmission electron microscope (JEOL JEM 2100 with a LaB6 cathode at 200 keV) was used to create the image of samples being deposited on C-coated Cu grids. | |
Results and Discussion |
|
| Figure 1 (right) shows the heterogeneous shape of quasi spherical monodisperse GNPs. The sedimentation (pressing) and collisions among the nanoparticles under the centrifugal force can be the cause for aggregation and therefore influence the binding activity of IgG. Apart from possible physical damages, multiple centrifugations steps of nanoparticles sum up to a time consuming process to prepare biocompatible nanoparticles. | |
| Figure 1: Representative signals of AIMS sum signal and humidity of the exhaled breath sample measured with ChemPro®100 and multicapillary column used. The AIMS spectrum from the maximum point of the AIMS sum signal was analyzed further. | |
| We used a new method to avoid multi step centrifugation of NPs for removing excess of chemicals or proteins [15]. This robust method consists of one or more simple reaction chambers with lateral filter membranes, which are permeable for molecules with defined sizes. At the bottom of each reaction chambers is a small well in the form of a pocket to collect modified NP (Figure 2A). The same method was used to separate small “native proteins” such as BSA (bovine serum albumin) from IgGs or unbound IgGs from colloidal solution. The following procedures describe the benefits of the novel reaction/separation chambers. Most manufacturers use proteins such as BSA, gelatin or sugars as stabilizer to protect their products (for example antibodies) against aggregation [16]. Such proteins are able to bond with NPs covalently, like IgGs or enzymes because of their free amine residues. Figure 2B demonstrates the separation of small “native proteins” (stabilizer) from IgG/stabilizer mixtures by this new method. The method is based on a continuous flow of buffer through three chambers. Chamber C1 contains the compound of IgGs and stabilizer at the beginning of the separation. In the first step the solution and both proteins move through the first membrane (pore size=150 kDa) under vacuum. In the next step only the stabilizer runs across the second membrane (cutoff of 100 kDa) of chamber C2. Thus in the end pure IgG molecules will be collected in chamber B whereas stabilizers such as BSA are discarded into the last chamber (C3). | |
| Figure 2: The reaction chamber is used for continuous conjugation of IgG onto NPs. The solution moves with excess of IgG through the membrane (pore size=150 kDa) and IgG conjugated NPs are collected in well (2A). Schematic description of protein separation by using three reaction/ separation chambers (2B). Chamber C1 contains the compound of IgG and stabilizer (red stars). Yellow dashed line between chamber C1 and C2 demonstrates the first lateral filter (cut off=150 kDa). IgG molecules are collected in chamber C2 and stabilizer molecules can move through second lateral filter (yellow dashed line) between chamber C2 and C3 (cut off=100 kDa). | |
| We used SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) and subsequently performed western blot analyses to examine the quality of the new separation method. The term electrophoresis refers to the movement of SDS-coated proteins in response to an electric field. Reducing agents such as ß-mercaptoethanol break up intra- and inter-molecular disulfide bonds of IgG (150 kDa). Therefore in the presence of ß-mercaptoethanol, two distinctive bands were observed by immunoblotting, corresponding to the molecular weights of heavy (50 kDa) and light chains (25 kDa) of antibodies [17]. We had no information about the type and amount of stabilizer in the purchased IgG/ stabilizer solution. Our determination of total amount of proteins by using UV-Vis spectroscopy (ultraviolet-visible spectrophotometry) shows 10:1 molar ratio of stabilizer to IgG (data not shown). | |
| Figure 3 shows the results of Ponceau S staining and western blot analysis of 1 μg commercial IgG/stabilizer before (C1) and after separation (C2 and C3). Due to low sensitivity and the detection limit for Ponceau S staining, no IgG bands (25 or 50 kDa) were seen on the membrane. Nevertheless the sensitivity was sufficient to show the band of the stabilizer (ca. 10 μg) in chamber C1 and C3. In contrast to Ponceau S, the two distinct bands of IgG could be detected by western blot analysis in chamber C1 and C2. The isolated anti p63 IgG from chamber C2 was then used for labeling of metallic gold/silver nanoshells (Au@Ag) in conjunction with SERS microscopy. We used tunable hollow Au@Ag nanospheres with a particle diameter of 55 nm and extinction maxima in the range of 600-700 nm as SERS substrate [4,12-14]. Figure S1 (left) shows SERS labeled NPs with a 100: 1 mixture of 2-nitro-5-thiobenzoate (4-NTB), and 16-Mercaptohexadecanoic acid (16-MHA). 4-NTB works as Raman reporter molecules in mixture and 16-MHA is a thiol-containing spacer molecule with terminal reactive group (COOH) for bioconjugation. Numerous protocols are available for bioconjugation of metallic NP [12-14,18]. A textbook example of bioconjugation chemistry is the activation of carboxylic acids by N-hydroxysuccinimide (NHS) and N-Ethyl- N-(3-dimethylaminopropyl) carbodiimid (EDC). After activation of the terminal COOH moieties of the longer 16-MHA spacers, the resulting NHS esters are conjugated to primary amines, for example lysine residues of enzymes or IgGs. The conjugation product, a SERSlabeled antibody, is shown in Figure S1(right). | |
| Figure 3: Left) Ponceau S staining of 1 µg commercial IgG/stabilizer (C1) in comparison to collected solution from chamber C2 and C3. Right) Western blot analysis of the same membrane. | |
| Figure S2 shows the correlation of colloidal aggregation and the amount of crosslinker during preparation by scanning extinction spectroscopy. We used 60 nm GNP to determine an optimum amount of zero length crosslinking reagents (NHS and EDC). Hence the narrow plasmon band of GNP shows the formation of aggregates being more sensitive than broad plasmon bands of Au@ Ag (Figure S3 & S4A) [12,13,18]. The peak plasmon resonance of GNP shifts to longer wavelengths and broadens as a consequence of increased aggregation. The appearance of longitudinal resonance mode as the second peak is a signal of a severe colloidal aggregation. All NHS/EDC activated samples in Figure S2 were conjugated with HRP (Horseradish peroxidase) labeled IgG. The average number of antibodies on each labeled NPs was determined using an enzymebased assay [13]. Based on our results, the enzymatic activities are the same and there is no difference between having optimal or excessive amount of crosslinker (data not shown). In other words the excess of crosslinker together with centrifugation increase the degree of aggregation. High stability of SERS labels before and after bioconjugation is a necessary prerequisite for their successful application in bioanalytical chemistry. By way of comparison the decrease of aggregation could be achieved the use of a vacuum system and therefore avoiding centrifugation steps. Figure S3 reveals extinction spectra of IgG conjugated SERS nanotags, which were prepared in reaction/separation chamber. The plasmon peak of GNP exhibits a red-shift from 540 nm to 544 nm after bioconjugation. For quantitative SERS microscopy, sensitive and robust SERS labels with high degree of scattering efficiency and reproducible signals are required. The optical properties of Au@Ag are determined by several parameters such as size and chemical composition [12]. The design employed in this study is based on nanoshells (ca. 55 nm) with tunable plasmon resonances, which are optimized for red laser excitation (633 nm see Figure S4A). A previous publication reported that Au@Ag particles increase SERS sensitivity roughly eightfold more than GNP with the same size [4,12]. The tunable plasmon of Au@Ag is important because red laser excitation minimizes cellular or tissue autofluorescence, which degrades the signal-to-background ratio and image contrast. Therefore we used Au@Ag as an efficient SERS substrate with tunable plasmon resonances in the red to nearinfrared SERS substrate (see Figure S4A) [4,12,13]. The labeled anti p63 Au@Ag are used for the localization of the basal cell protein p63 in healthy prostate tissue [13,14,19]. The selective localization of nanotags depends only on the affinity of their binding ligands (IgG) to targets. Previous studies have shown that the formation of antigen-antibody complexes can be markedly affected due to changing of pH, ionic strength and temperature [20]. These factors have an effect on the binding affinity of antibodies to antigens and may cause both false positive and false negative reactions. The SERS false color images in Figure 4 are based on the intensity of the Raman marker band of 4-NTB at about 1340 cm-1 (Figure S4A). For the next step we prepared anti p63 coupled SERS probes with and without the use of reaction/ separation chamber. The p63 IgG labeled SERS probe in Figure 4 left has been prepared by use of the centrifuge. In contrast, SERS probe in Figure 4 right was prepared with the filtration/reaction chamber with lateral filter. The nonspecific binding of nanoparticles between the nuclei are clearly visible in Figure 4 left. On the contrary, Figure 4 right, shows only the labeled nuclei of cells with a diameter of about 6 μM [13,14,19,21,22]. The spaces between the nuclei of cells are not marked with SERS label. The improvement of biocompatibility of nanoparticles could only be achieved by using the reaction/separation chamber. We performed negative control experiments employing BSA-SERS nanotags conjugates, i.e., without antibodies. Negative control experiments with SERS labeled BSA indicate no or only minimal non-specific binding (Figure S5). | |
| Figure 4: Immunohistochemistry on prostate tissue from healthy donors using Au@Ag conjugated to an antibody against p63 proteins. The SERS false color images are based on the intensity of the Raman marker band of 4-NTB at about 1340 cm-1 (see Figure S4). The overlaid SERS false color images, demonstrating the selective abundance of p63 only in the basal cells (B), of the epithelium (E), stroma (S) and non-basal epithelial cells of prostatic gland; (L). Figure 4 left shows the non-specific binding of the antibody-conjugated nanoparticles, caused by centrifugation steps. Figure 4 Right: labeling of p63-proteins with nanoparticles, which were prepared in reaction/separation chamber. | |
Conclusion |
|
| This study has shown that the stability and biofunction of NPs depend on the amount of crosslinker and avoidance of centrifugation steps (Figure S2). The excess amount of crosslinker can cause aggregation of NPs which does not improve the quality of bioconjugation. The robust reaction/separation chamber with lateral filter membranes is used to separate stabilizer molecules from commercial IgGs solution. The same chamber is used for continuous modification and covalent coupling of IgG molecules onto various NPs. The excess of chemicals and proteins is removed from NPs without any centrifugation step. The formation of aggregation and nonspecific binding of SERS tags could be minimized by using separation chambers (Figure S3). As we have shown before, the staining quality in SERS microscopy depends, similar to standard methods, on several factors such as blocking buffers, antigen retrieval, incubation time, and amount of nanotags [22]. In this study we could introduce a novel method to protect the function of conjugated IgG. The tumor suppressor p63 is solely abundant in the basal cells of healthy donors but not in the stroma, secretory epithelium and lumen [13,19,21]. | |
| Data in Figure 4 demonstrates the selective binding of two differently prepared nanotags. The blocking buffers, antigen retrieval, incubation time, and amount of nanotags were the same for both samples. The only difference between these experiments was centrifugation steps to remove the excess of chemicals or unbound proteins such as IgGs. Only IgG conjugated nanotags, which were prepared in the reaction/separation chamber indicate minimized or absent non-specific binding. | |
Supporting Information |
|
| Supporting Information is available from the SciTechnol Library or from the author. | |
RAcknowledgments |
|
| We acknowledge financial support from University of Applied Science Osnabrück (KST, 58210045). The authors wish to thank Professor Michael Hensel (University of Osnabrück, Germany) for generous material support. | |
References |
|
|
|