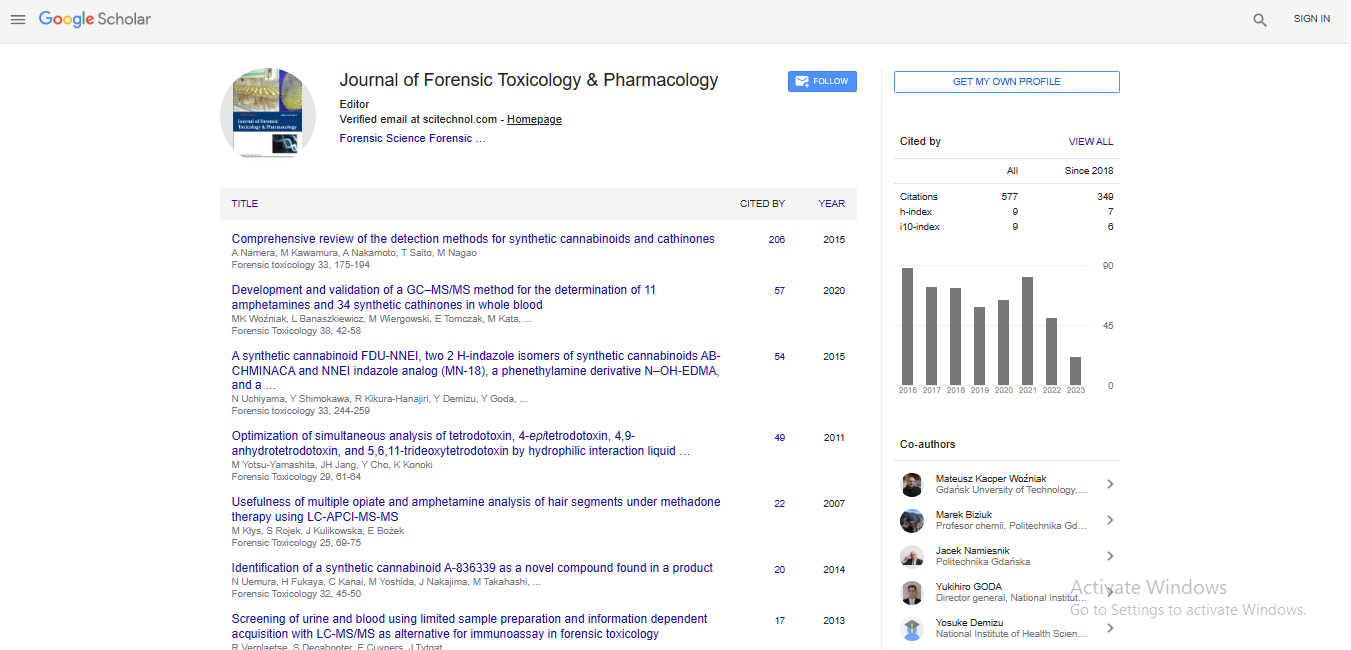
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Review Article, J Forensic Toxicol Pharmacol Vol: 3 Issue: 4
Hypothalamic Pathophysiology in the Neuroimmune,Dysmetabolic and Longevity Complications of Chronic Opiate Dependency
| Albert Stuart Reece*, and Gary Kenneth Hulse | |
| School of Psychiatry and Clinical Neurosciences, University of Western Australia, Australia | |
| Corresponding author : Dr. Albert Stuart Reece School of Psychiatry and Clinical Neurosciences, University of Western Australia, 39 Gladstone Rd,Highgate Hill, Brisbane, Queensland, Australia Tel: +617 3844-4000; Fax: (617) 3844-4015 E-mail: sreece@bigpond.net.au |
|
| Received: May 15, 2014 Accepted: June 30, 2014 Published: July 07, 2014 | |
| Citation: Reece AS, Hulse GK (2014) Hypothalamic Pathophysiology in the Neuroimmune, Dysmetabolic and Longevity Complications of Chronic Opiate Dependency. J Forensic Toxicol Pharmacol 3:3. doi:10.4172/2325-9841.1000126 |
Abstract
Hypothalamic Pathophysiology in the Neuroimmune,Dysmetabolic and Longevity Complications of Chronic Opiate Dependency
New conceptual and therapeutic advances in our understanding of various important pathophysiological states have remarkable relevance to current practices in the area of the treatment of opiate dependence. Major breakthroughs have recently occurred in our understanding of: The central mediation of the hypertensive-obesity-metabolic syndrome and particularly its association with the hypothalamic regulation of longevity; The ability of addictive chemical species to induce local inflammation of many parts of the CNS including the hypothalamus; The interactions between the host and the resident gut microbial flora and its implications for systemic organismal health and disease. These breakthroughs have occurred on the backdrop of detailed quantified pathophysiological and mortality surveys of opiate dependence from several continents and in the context of exciting new pharmacological developments including suppressors of central neuroinflammation such as ibudilast, non-habituating newer generation opiates such as PTI-609, and depot-implantable forms of the opiate antagonist naltrexone. Opiates induce electrical silencing of POMC neurons resulting in hyperphagic obesity and dysmetabolic syndrome, mimicking senescence. Hence POMC neuronal pathophysiology becomes amplified organism-wide in a feed-forward loop. Together these factors suggest that we are on the very threshold of new insights which have the potential to overhaul both our conceptual understandings and our clinical treatments. The purpose of this review is to bring together findings from widely disparate areas and to elucidate their relevance and overlap with research on the effects of long-term opiate dependence. Whilst opiate dependence is the primary focus these observations likely also pertain to other chemical dependencies.
Keywords: Opiates; Addictive drugs; Metabolic syndrome; Ageing; Longevity; Central neuroinflammation; Stem cells; Peripheral and central stem cell- immune interactions; Morphine; Heroin; Buprenorphine; Methadone;
Keywords |
|
| Opiates; Addictive drugs; Metabolic syndrome; Ageing; Longevity; Central neuroinflammation; Stem cells; Peripheral and central stem cell- immune interactions; Morphine; Heroin; Methadone; Buprenorphine | |
Introduction |
|
| Classical studies have shown that patients dependent on a variety of addictive agents harbour a wide variety of associated pathological features [1]. Large epidemiological studies conducted over many decades from respected groups confirm greatly elevated rates of death amongst opiate dependent patients from disease and dysfunction of all major organ systems [2] and an increased number of relative years of life lost [3]. Elevated rates of death from tobacco, alcohol and stimulant use are also widely acknowledged in the literature [4-6]. A series of papers from our group and others has shown that opiate dependent patients demonstrate elevated rates of clinical (dental [7-9], psychological [10,11], hair greying [12,13], cancer formation [2,3,14,15]) and laboratory (alanine aminotransferase, high sensitivity C-reactive protein, erythrocyte sedimentation rate, reduced circulating stem cells [16-18]) biomarkers of the ageing process, and indeed manifest an increased need for specialist geriatric care at early ages [11]. It has recently been shown in both sexes in crosssectional and longitudinal studies that lifetime opiate dependence is dose-dependently characterized by increased rates of arterial stiffness, a major measure of cardiovascular age, and an important surrogate for organismal age [19-22]. These epidemiological, clinical and laboratory results quantify and provide evidentiary weight to the classical observation of accelerated ageing in opiate dependent patients made as long ago as 1899 [23], which can reasonably be extended to other common forms of chemical dependency. | |
| The toxicological manifestations of opiate and drug dependency suggest that more diverse biological pathways may be at play beyond the strictly receptor-ligand interactions at classical opiate receptors of the mesocortical limbic system conceptualizations which usually dominate mechanistic studies in the field [24]. Indeed classical opiate receptors have been found to comprise only 2% of the saturable opiate binding in several brain regions [25]. Other authors have written of the effect of opiates to impair stem cell activity via a non-classical cytoplasmic -perinuclear enkephalin receptor [26] and stimulate the innate immune system via the toll-like receptor 4 (TLR4) – myeloid differentiation factor 2 (MD2) heterodimer [25]. Importantly stem cells in most tissue beds are highly sensitive to the interactive negative regulation by immune activity [27-32], and the systemic actions of this trilogy of effects have been have been noted previously in the addiction literature [21,33-36]. | |
| Recent conceptual advances however have moved this well appreciated focus from the whole organism to the brain and particularly the hypothalamus. One simplified depiction illustrating hypothalamic anatomy is shown in Figure 1 (from http://www.cellbiol.net). Important parallels exist between recent advances in obesity research and drug addiction. Both syndromes represent clinical scenarios of the dysregulation of appetite, one for food and the other for drugs of dependence. Both are characterized by low grade inflammation in the brain. In the case of obesity the low grade inflammation is seen primarily in the mediobasal hypothalamus (MBH), particularly in the paraventricular and acuate nuclei. Importantly these two nuclei are home to two important clusters of neurons, the first being anorexigenic and elaborating the endorphin precursor proopioiomelanocortin (POMC) and cocaine-amphetamine transcript (CART), and also the orexigenic neuropeptide Y (NpY) – Agouti Related protein (AGRP) neurons [37]. The opposing effects of these two sets of neurons in the mediobasal hypothalamus are accepted as directing appetite regulation across the body. The hypothalamic POMC neurons have a principal determinative action in inhibiting appetitive and acquisitive behaviours relating to food and other pleasurable stimuli, likely including other drug use. Hypothalamic networks have diffuse connections to many other areas of the brain including the sympatho adrenal system and can trigger appropriate feeding or satiety behaviours. Importantly opiate agonists have been shown to hyperpolarize POMC neurons within seconds [38] and opiate antagonists have been shown to depolarize them [39], which doubtless underlie the clinical observations relating to a preference for foods high in saturated fats and simple carbohydrates and increases both of body mass and total body fat and increased drug use by patients treated with opiates, and reduced appetite for food [39] and other addictive drug use [40,41] by patients treated with antagonists as has been previously documented [34,42]. | |
| Figure 1: Hypothalamus Location and General Anatomy. | |
| The fact that POMC neurons also elaborate CART, and receive (sexually dimorphic) inputs from endocannabinoid [43-45] and endogenous benzodiazepine / GABA receptors [38] and stimulatory cholinergic neurones implies that these critical neurometabolic cells are able to integrate information related to the most commonly used addictive chemical species including tobacco, stimulants such as cocaine and amphetamine, benzodiazepines, alcohol and cannabinoids. This in turn suggests that many of the conclusions made in this paper arising from considerations of the pathophysiology of opiates in fact carry boarder implications - with some obvious modifications - for the gamut of addictive agents commonly encountered clinically. | |
| Very excitingly recent advances in the field of obesity research have identified that low grade mediobasal hypothalamic neuroinflammation is the driver of not only appetite dysregulation, but the whole clinical panorama of the metabolic syndrome (including obesity, hyperlipidaemia, insulin resistance, hyperglycaemia, type two diabetes, leptin resistance, reduced activity, essential hypertension, reduced thermogenesis), all of the features of which have now been described in opiate dependence. Moreover as the functions and activities of this key regulatory area are increasingly explored, so too the implications of serious dysregulation in this area become increasingly profound, and explicate in more detail the previously unappreciated interrelationships between many of the diverse and seemingly disparate clinical features of the syndrome of long term opiate dependence. | |
| Part of the excitement derives from the increasingly wide functional implications of this hypothalamic locus. Whilst it has long been recognized to be involved with neuroendocrine regulation as the “master regulator of the endocrine orchestra”, the activities of these hypothalamic nuclei now include also blood pressure regulation, control of autonomic tone, appetite and feeding regulation, glucostat function, lipostat function, circulating amino acid control, body weight control and thermogenesis [46-50]. The arcuate and paraventricular nuclei also have close links with the hypothalamic supraoptic nuclei which is known to be the master regulator of the short term time cycles in the body of the circadian rhythm [51-55]. Moreover modulation of the circadian rhythm has been shown to impact the metabolic derangements of the metabolic syndrome and induce weight loss [56-58]. Of key importance the arcuate nucleus and its homologues have been shown to exert an important regulatory control over organismal lifespan determination in flies, roundworms and mice [49,59-64]. | |
| For these reasons the implications of hypothalamic neuroinflammation as has been described in opiate, alcohol, tobacco, amphetamine and cocaine dependence are even more far-reaching than have been previously characterized. Whist the trilogy of stem cell inhibition, inflammation, and the interactive effects of inflammationassociated suppression of stem cell activity have been noted to act systemically [9,17,33,35,65], the fact that they are also clearly acting in the hypothalamic control centre whose metabolic regulatory functions are being increasingly uncovered greatly amplifies the significance of these effects. Indeed it has been shown that in a model of metabolic derangement induced by high fat feeding, hypothalamic inflammation occurs within just a few days, and substantially precedes that of the remainder of the organism [66]. | |
| It is not suggested that these effects occur in isolation from other systemic effects of opiate dependence. For example it is well documented that the administration opiates tends to increase preference for fatty foods and foods high in simple carbohydrates [37,67-73]. Such changes will clearly exacerbate their centrally mediated anti-lipostatic and anti-glucostatic functions [74,75]. Similarly immune activation can increase the leakiness of the gut mucosal barrier to enteric commensals, increase hepatic damage and scarring, and by increasing shunting through the liver increase the leakiness of the liver barrier to systemic endotoxaemia [76-78], further exacerbating the immunostimulatory changes of chronic immune over-stimulation. These effects will be exacerbated by the well described constipating effects of opiates, an effect which is commonly problematic in the clinical management of opiate dependency, and to which habituation does not occur [79,80]. Immune compromise in the oral-dental mucosa implies that in many patients poor oral hygiene and advanced dental decay and chronic periodontitis also contributes to the subacute and chronic release of bacterial antigens and organisms into the general circulation [7,8,81,82], further becoming a major source of highly potent and toxic TLR4 ligands, and a long term source of immune stimulation [83-92] and thus immunometabolic dysregulation [93]. | |
| The importance of enhancing our understanding of the medical pathophysiology underlying chronic opiate dependence was recently underscored by a detailed review of the world’s largest cohort, from New South Wales in Australia [94]. This study examined 43,789 patients over 412,216 patient years in the years 1985-2005. This study noted that as the mean age of opiate dependent patients in many nations is rising, and that as their age rises, the relative importance of degenerative age related disease rises, and particularly for cardiovascular disease, cancer and liver disease. Indeed in this study over half (50.5%) of the years of life lost, which averaged 44 years per patient, we related to deaths not attributable to opiate overdose, and thus were due to medical causes. Another study which specifically reviewed larger series focussing on older opiate dependent patients noted a prevalence of chronic medical disorders of up to 89%, hepatitis C rates of up to 94%, hypertension rates of up to 58%, heart disease up to 53% and hyperglycaemia and diabetes rates of up to 18% [95]. | |
| Whilst all these effects are important and significant, the focus of the present article will be to review recent developments in obesity related neuroinflammation as a model for the low grade brain inflammation seen in opiate dependence, and consider their implications for the clinical spectrum of disease to which patients and their attending clinicians are exposed. Whilst opiate dependence is taken as the paradigmatic model for the purposes of this discussion, in fact many of the comments made here relate with minor modifications to other chemical dependency syndromes as addictive chemicals generally induce central neuroinflammation [25,93]. | |
Pathology of Opiate Dependence |
|
| Table lists out various selected pathologies described in opiate dependence outside the neuraxis (Table 1). For many decades it has been known that opiate dependence is related to much different pathology found at post mortem [1]. However recent quantitative studies in the last decade have allowed more precise quantification of these effects, in many cases compared to population means and norms. The first column of the table lists the condition under consideration by organ system. The central column shows the effect size which has been described together with the applicable references, and the third column shows the association of the various pathologies with described changes of ageing. Aging too has numerous pathological expressions. For convenience the tabulation of the effects of ageing provided by Lombard et al., in 2005 has been used as a reference point [96]. The major sources of data for the table are Darke [97,98], Degenhardt [2], Hser [3], Khademi [14] and Rosen [11] from Australia, California, Iran and the USA respectively. The studies from both New South Wales [2] and Golestan [14] are particularly notable for being of unusually large size comprising some 425,998 and 234,928 person-years of follow-up respectively, and therefore having unusually robust findings. The extraordinarily close quantitative agreement between the major findings of these two studies is at once both astonishing and compelling. Such close agreement in relative risks would appear to be most unusual in the epidemiological literature. | |
| Table 1: Selected Quantitative Systemic Pathophysiology of Opiate Dependence Cf. Ageing Pathologies in Lombard [96]. | |
| Numerous fascinating points of interest emerge from this table. One clear point of interest is the overall commonality of the pathologies of addiction with many of the described changes seen in ageing. It is understood that were more complete list of the pathologies of ageing to have been employed, more overlap and intersection would have been noted. | |
| Secondly whilst it is well known that stimulant abuse of agents such as cocaine and amphetamines is associated with high rates of cardiovascular disease, this is also true of opiates. In fact this is a very robust finding and was found particularly by the work of Darke [97,98], Degenhardt [2], Hser [3], and Khademi [14] and was noted by Khademi to occur in both sexes. Moreover the effect size is clearly substantial so that Hser found cardiac death (measured as years of potential life lost) to occur at a rate 3.2-fold that of the general community, and ventricular hypertrophy was noted in 10.7% of opiate dependent decedents over 44 years of age by Darke [98]. All the cardiac pathologies of the first section of Table 1 were 2-4 fold worse in methadone compared to heroin dependent clients, consistent with their likely greater opiate exposure [97]. Acute pulmonary oedema is believed to be a common terminal event at death in opiate dependence. | |
| Similar results have been noted with severe coronary atherosclerotic disease with odds or hazard ratios of 2-4 times controls in studies from Australia and various parts of Iran [14,99,100]. Both hypertension [11,74] and elevated C-reactive protein [16-18] have also been noted. Both conditions are known to have extensive links with cardiovascular disease. It is therefore not surprising that elevated rates of cardiovascular death have been found. The very close agreement between the rates of elevation of the rates of cardiac death in the two very large studies, namely 2.20 [2] and 1.81 [14] is also most remarkable. The first measure is a standardized mortality ratio and the second is a hazard ratio [14]. In that they originate from very large population controlled cohorts, each ratio is likely to be internally robust. That these accord so closely with each other with significant overlap in the 95% confidence intervals (1.9-2.5 and 1.56-2.09), also strongly suggests external validity and reliability. Whilst there is a well known discourse related to the prolongation of the QTc interval by high dose opiate agonists [101-108], there is little discussion of this topic outside of this context. The authors have conducted over 15,000 individual pulse wave analysis studies [19-22,36] during which the very clear view was formed that cardiac arrhythmia was much more common in the opiate dependent group (ASR, GKH unpublished data). | |
| Similarly respiratory disease and gastrointestinal disease is also noted to be elevated amongst opiate dependent patients, as documented by several sources [2,97]. | |
| A large literature documents the very elevated rate of neuropsychiatric disease in many chemical dependencies including opiate dependence. Depression and anxiety are two of the most common manifestations of psychiatric distress in addicted patients with some clinical series reporting rates of 60-80% [11,109], and are also well described in the ageing literature. Stroke has also been found to be more common in opiate dependence. Suicide is a terminal event in depression. Anxiety and depression are now believed to have important inflammatory neuropathological substrates, and this important development is discussed further in the following section. As indicated in Table 1 brain atrophy is also well described in opiate dependence, and is also well known to occur at increased frequency in ageing. Moreover one recent study has shown a direct correlation between a reduction in telomerase activity of circulating leukocytes, brain structural atrophic changes in grey and white matter, and reduced executive functioning, memory and attentional control on fMRI studies even as long as one year after heroin abstinence [110]. | |
| Both haematological [74,79,111-113] and orthopaedic [112,114-118] complications of opiate dependence are described, and are known to occur with increased frequency with age. | |
| The subject of the immunological changes seen in opiate dependency is a very large one. Signs of both immune stimulation and suppression have been noted [79,93,119]. Recent evidence suggests that the immunosuppression is an expression of the immune stimulation, such that what is in fact observed is an immune dysregulation as partially homeostatic mechanisms attempt to compensate for numerous pharmacologically induced modulations [93]. Numerous stigmata of immune stimulation have been described as occurring in opiate dependency which closely parallel the age-related “inflamm-ageing” well described in the ageing literature [120-123]. Moreover it is well established that amongst human nonagenarians, those with serological and cytokine evidence of diffuse immune stimulation face a 60% mortality rate, which is not shared by those whose immune system is quiescent [122,124]. Clearly this paradigm, which is referred to in Goodman and Gilman’s text as polyclonal gammopathy associated with chronic methadone use [79], in patients of very advanced age carries numerous similarities with our much younger patients with the immune stimulation / suppression pattern characteristic of drug dependency. In that drugs of abuse commonly cause immune stimulation this is a very important theme which is developed at some length in following sections. Moreover there is now a well-established link between immune stimulation and cancer induction in many tissue beds including the skin, bowel, liver parenchyma, oesophagus, stomach, bronchus and lung and biliary tree [4]. As noted below, increased rates of cancer are seen both in ageing and opiate dependency. | |
| Numerous endocrinopathies have been described in opiate dependence which are summarized in this table and elsewhere [75,125]. Importantly many of these endocrine systems are directed through the hypothalamic-pituitary axis, which as documented below is significantly perturbed in diverse dependency syndromes. Interestingly although the literature is divided on the point, several reports exist that the hypothalamic-pituitary-adrenal, hypothalamicpituitary- thyroid and hypothalamic-pituitary-gonadal axes are relatively suppressed either in absolute terms (low circulating levels of the end organ hormone) or functional terms in terms of response to stress of the axis of control [79,126,127]. One of the most important of these disorders and also one of the least known, is the 3-4 fold elevation in rates of diabetes mellitus seen in opiate dependent populations [75,111,128-132], along with the complex of the metabolic syndrome which frequently accompanies it including hyperlipidaemia, hypertension, central adiposity and hyperfibrinogenaemia [74,75,111,128,130]. Data from one such recent study [75] which documents the three week and three month average blood sugar results as fructosamine and HbA1C respectively is re-presented in Figure 2. Also of considerable note is hypogonadotrophic hypogonadism which has been noted in opiate dependence in both sexes and is associated in females with amenorrhoea, oligomenorrhoea and reduced fertility, and in males with erectile dysfunction, reduced libido, lowered sperm count and osteoporosis [37,79,112,133-134]. Whilst the prime focus of the research to date has been on the effects of peripheral hypogonadism, it may be that the centrally mediated hypogonadotrophism is at least as important a part of this pathophysiological perturbation as suggested by recent research [59] and discussed further in Sections III and V. | |
| Figure 2: Glycaemic parameters by age, addictive status and sex. | |
| Advanced dental pathologies are both a sign and a cause of age-related change [7-9]. Both antigenic and viable microbial material has been detected in distant endovascular sites particularly in luminal plaque, which clearly form a powerful and chronic immune stimulus. Advanced dental decay commonly contains many gram negative anaerobic and microaerophilic bacteria, such as Porphyromonasgingivalis, whose cell walls are powerful toll-like receptor (TLR) ligands, particularly at TLR’s 4 and 2. “Growing long in the tooth” which corresponds to the clinical diagnosis of chronic periodontitis, is of course one of the primary stigmata of human ageing [135,136]. | |
| A large literature documents the effect of opiates to increase the preference for unhealthy diets high in simple carbohydrates and saturated fats [38], which is often accompanied either by overt increases in body weight [137], or increases in body fat content, with corresponding reductions in body protein so that overall weight is little changed. Similarly many studies confirm high rates of tobacco, alcohol and other drug use in opiate dependent populations. The pattern of other drug use is generally determined by local availabilities. These dietary and consumptive changes will likely compound the other elevated cardiovascular risk factors noted above. | |
| Several studies show elevated rates of genitourinary disease in opiate dependence which is again a well-established finding in the ageing literature. | |
| Cancer is one of the major features of advancing age in many mammalian species. Indeed with the exception of cancers related to congenital abnormalities cancer is considered to be an age-related disorder. The estimates of increased cancer incidence in opiate dependence in the Sydney study was 1.6 (95% C.I. 1.49-1.9) [2], in Iran 1.61 (1.28-2.03) [14] and the years of life lost in California due to cancer was said to be 2.05 times that of the remainder of the US population [3]. Clearly these results are in extraordinarily close agreement. | |
| As shown in Table specific cancers have been found to be elevated in opiate dependency (Tables 1,2). The Golestan study found elevated risks of all tumours, lung in men, and “other” tumours, but not oesophagus or stomach [14]. This subject was considered in detail in a follow-up report from the Sydney group using a somewhat extended database incorporating 481,936 person-years of follow-up with a mean age at treatment entry of 27 years, and a mean time of opiate substitution treatment of 2.6 years [138]. Ten cancers were noted to occur at elevated rates (Kaposi’s sarcoma, liver, bronchus and lung, larynx, vulva, anus, tonsil, pancreas, unknown primary and cervix), and 6 at reduced rates (melanoma, colon, brain, breast, thyroid and prostate). In relation to prostatic cancer, generally a tumour of advanced age, the relatively young age of the cohort is relevant. Exposure to viral risk factors such HIV, Hepatitis B and C and human papillomavirus were shown statistically or believed to be a signficant compounding risk factor. Tumour occurrence was almost completely confined to those co-infected with Hepatitis B, C or HIV. The average age standardized percent change increased dramatically over the 22 year observation period (1985-2007) of the study by 9.4% [138]. | |
| Table 2: Mechanisms and Cell Types of Central Neuroimmune Stimulation in Opiate Dependence - After Hutchinson (2011) [25]. | |
| This work was supported by a variety of studies largely from northwestern Iran where the consumption of opium by smoking is widespread. Geographically Iran is contiguous with Afghanistan, the source of about 85% of the world’s opium poppy supply [139]. Disease specific studies exist of elevated rates of bladder, oesophagus, oral, laryngeal and lung cancer [140-148]. Striking findings were noted in studies of bladder cancer with odds ratios of 7.49 (95% C.I. 1.33- 159.53) calculated from some studies [140], and in laryngeal cancer where odds ratios of 25 were noted, with effects robust to adjustment for tobacco consumption [144]. In one large study, the crude odds ratio for laryngeal cancer in opium exposed patients was 21.55 (C.I. 10.5-44), which fell to 10.74 (5.7-2.0) after adjustjent for tobacco [144]. Similarly in a recent large study of lung cancer the tobacco (and other covariate) -adjusted odds ratio for opium exposure was 3.1 (1.2-8.1), and when co-exposed in combination with tobacco, the odds ratio rose to 35.0 (11.4-107.9) [148]. | |
| Abnormal pain states including hyperalgesia (heightened pain experienced from noxious stimuli) and allodynia (pain experienced from non-painful stimuli) are well described in opiate dependent patients consistent with the centrality of opioids including endorphins and enkephalins to the pain perception system. | |
| A very wide variety of neuropathologies has been reported in opiate dependence and the interested reader is referred to various specialist reports of these findings [24,149-152]. Whilst there is significant divergence in the literature, there are several reports of reduced grey and white matter and or demyelination associated with opioid dependency, some of which seem to be related to changes of acute or chronic hypoxia [150,151]. The globuspallidus, basal ganglia and hippocampus are particularly affected, with one large postmortem series of 180 cases reporting that 80-89% of cases examined had molecular changes of astrocytic (glial fibrillary acid protein) or microglial (CD68) activation [151,152]. The hippocampus is closely related both structurally and functionally to the hypothalamus. The basal ganglia are also located close by. Interestingly dramatic increases in the products of lipid peroxidation (malondialdehyde) to almost twice the control level have also been shown both in cortex and white matter [153] and in isolated astrocytes [154]. Stroke, perivascular bleeding, hypoxic-ischaemic leukoencephalopathy, nerve cell loss, numerous brain infections, myelopathy, terminal cerebral oedema and spongiform leukoencephalopathy have also been reported [150,152]. | |
| The question arises as to the findings of hypothalamic inflammation in post mortem series of opioid overdoses. Unfortunately it has not been possible to locate studies which define this feature exactly despite search of PubMed, Google Scholar, PsychInfo, Embase and Ebsco Host did not reveal any such studies. Detailed search of several leading data sources such an authoritative monograph on drug overdoses [155], several detailed pathology reports of opioid overdoses [97,98,156], the landmark nine major US cites pathological survey reported by NIDA [1], specialist neuropathology articles [149-153] and careful large US pathological surveys [3] were uniformly unrevealing. | |
| As is made clear by the group of Hutchinson and Watkins all these neuropathological changes are compounded by the use of multiple addictive drugs, which in general not only tend to cause similar neuroinflammatory changes, but also tend to exacerbate each other [25,93]. The same authors list the hypothalamus as also involved in the changes in the neuraxis induced by the drugs of addiction. | |
| As noted above, many age-related pathologies are elevated in opiate dependence including grey hair [12], chronic periodontitis [8], reduced circulating stem cells [65], osteoporosis [112], neuropsychiatric disorders [4], cancer [2,3,14,138], skin problems, abscesses and ulcers [113,157,158], polyclonal gammopathy [79], and disturbance or inversion of the circadian rhythm [79]. The elegant work of the Taiwanese group who directly tested this hypothesis is especially noteworthy in this regard [110]. These workers found an 88% reduction in peripheral blood leukocyte telomerase activity of opiate users even after one year’s sobriety, and found significant interactions between heroin use and prefrontal cortex damage to grey and white matter, and memory, attentional and executive functions of the brain at fMRI [110]. | |
| Multisystem disease has been noted to occur in 44% of patients over the age of 44 years [98]. Multisystem disease has been noted to occur 3.3 times more often in methadone-exposed as compared to heroin-exposed decedents [97]. Similarly any system disease has been noted to be more common in methadone- as compared to heroin- exposed decedents with an odds ratio of 3.4 [97]. | |
| One of the major results to emerge particularly from the largest studies in this area, that of Degenhardt and colleagues in their Supplementary Appendix 6 [2] and of Khademi and colleagues in their Table 3 [14], is that across all organ systems examined the rates of death are elevated, and particularly in the case of the former study [2], generally very dramatically so. | |
| The literature relating to neonatal and developmental outcomes following intrauterine exposure is large, complex and often contradictory [159,160]. There is agreement that rates of neonatal abstinence syndrome seen in babies born after maternal opioid treatment are significant, although the long term health implications of this are unclear [161,162]. | |
| Many studies have found that various congenital defects are more common in opiate exposed infants than in the general population. Reductions in birth weight, head circumference, and length have been noted as have several heart defects, gastroschisis, spina bifida, hydrocephaly and glaucoma [163]. Whilst several authors have found that prematurity rates, foetal size or head circumference is not altered in methadone exposed infants, many workers find that the incidence of small for dates, prematurity, and reduced head circumference is seen at higher rates or with statistically significant elevations in frequency [159-161,164,165]. Furthermore some [166] but not all [160] investigators were able to demonstrate a dose-response relationship between pregnancy methadone dose and increase of infant body size reduction, hospital stay and prematurity risk. One such study showed a reduction in infant mean head circumference at birth from 34.7cm to 32.2 cm [164]. This is equivalent to a 20.1% reduction in skull volume from 705.5cm3 to 563.7 cm3 (assuming perfect sphericity). One large study found a significant effect of opiates on birth weight, but this disappeared after multivariate adjustment for various factors including both weight [167]. Since birth weight is known to be related to opiate exposure this would appear to be a case of statistical over-correction. A similar large study used a similar multivariate technique [168]. These studies both controlled for other drug utilization. Whilst from a toxicological point of view this is appropriate, in that opioids clearly increase the hedonic drive and the consumption of pleasurable substances, this is also likely to be in some senses a case of over-correction. | |
| Buprenorphine is a partial opiate agonist. One careful study from Johns Hopkins Hospital and the National Institute of Drug Abuse found a close correlation between buprenorphine meconium concentrations and reduction in foetal head circumference (R = 0.874, P = 0.01), and a borderline association with reduced overall foetal length (P = 0.06) [169]. Another study showed a lower prematurity, small body size, head circumference, neonatal abstinence syndrome and gestational age in buprenorphine exposed compared to methadone exposed babies [170]. | |
| Another study found an increased rate of feminine behaviours in boys who had been exposed to opiates in utero, including crossdressing, female impersonation, drama improvisations, and female interactions [171]. No such effect was seen amongst methadone exposed girls. One Australian study of 22 infants exposed to methadone in utero found prolongation of visually evoked potential latencies which remained after multivariate adjustment [161]. | |
| One important longitudinal study found lower scores on the Bayley Scale of Infant Development in methadone exposed babies at 12, 18 and 24 months [165], although it has been reported that this effect disappeared after multivariate adjustment. Indeed in the poorest performing group in this study at 36 months 85% (3/21) of the babies were from methadone exposed mothers. When the children were studied at 84 months there was a higher percentage of abnormalities of gross and fine motor incoordination, poor balance, decreased attention span, hyperactivity, and language and speech delays. Academic and behavioural problems were also an issue, with little improvement from 36 to 84 months, and little flexibility in their developmental course. | |
| In this connection it is very important to note that several molecules which are usually considered to be immune active signalling molecules have a key role in determining neuronal structure, architecture or dendrite pruning. This includes TNFα (tumour necrosis factor-α), TGFβ (transforming growth factor-β), IL-6 (interleukin-6), MHC (Major Histocompatibility Class) class I molecules and their receptors and receptor components, DSCAM (Down’s Syndrome Cell Adhesion Molecule) families, neuronal pentraxins, and C1q and C3 complement fragments [30,172]. Moreover the toll-like receptors, around which much discussion centres in subsequent sections, were so named for their similarities to the toll receptors of drosophila. The drosophila toll receptors were actually first identified as major morphogens for their role in body pattern dorso-ventral polarity formation, cellular adhesion contacts, CNS midline determination and guidance, and the folding and formation of invaginations in the aero-oropharynx and around the anogenital grove [173-177], eye formation [178], oocyte orientation [179], and for motoneuron and muscle development [180]. Immune stimulation has also been shown to lead to foetal brain histological abnormalities, and learning and behaviour defects in adult life [181] and chronic neurodegeneration [182,183]. It is therefore not surprising that several major developmental studies of infants exposed to opiates in utero have consistently found significant behavioural and learning difficulties occurring with higher frequency and severity [164,165,184,185]. | |
| Moreover, GABA, is one of the major inhibitory neurotransmitters of the adult CNS, is stimulatory in the foetus developing in utero, and is critically involved in synaptic formation, cell proliferation, neuronal migration and dendritic maturation [159]. GABA is known to be important in memory formation and consolidation and in the regulation of fear, stress, and anxiety [159]. Moreover GABAergic interneurons form one of the major inhibitory inputs onto dopamine neurons, and inhibitory inputs from MOR-responsive opioidergic neurons regulate GABA and thus dopamine neurons. Thus opioids act to inhibit GABA and increase limbic dopamine [159]. Hence these systems play an important role in brain morphogenesis also acting in their role as neurotransmitters. | |
| A detailed review of the fascinating literature relating to the involvement of the opioid system in autism spectrum disorders (ASD) is beyond the scope of the present review. Core features of ASD involve deficits in social and communication processing [186]. However a number of fascinating observations contextualize and enlarge the present discussion. As long ago as 1978 Panskepp described the importance of the opioid system in social interactions [187], although the endocannabinoid and dopaminergic systems are also known to be involved [188]. This has been shown to apply to many situations including attachment between the rodent dam and her pups, adolescent play, and sexual encounters [189]. These findings have since been generalized to many species including the mouse, rat, chicken and primates [188]. It has been shown that this effect is mediated by the μ-opioid receptor (MOR) and is localized in the shell and core of the nucleus accumbens, a key node in the limbiccortical circuits concerned with brain reward [188]. Exogenous opioids (endorphins), exogenous opioids (exorphins, often derived from foods including milk and flour) [190,191] and pharmacological opioids predictably enhance these pro-social effects, while opiate antagonists reduce them [188,192]. Similarly genetic downregulation of MOR activity in the MOR-KO mouse has been shown to increase stress and anxiety behaviours, and this has been shown to be rescued by treatment with an mGlu4 agonist [189]. Indeed the MOR-KO mouse forms an excellent monogenic model for ASD [186,193]. Interestingly MOR-KO was also associated with major changes in the stimulatory glutamate and inhibitory glycinergic and GABAergic signalling and with changes in oxytocin and CRH (corticotrophin releasing peptide) levels [189]. Indeed immediate early gene activation (Fos and Egr1) fell dramatically, and oxytocin down-regulation in the nucleus accumbens fell by a factor of 12-fold. | |
| A recent systematic review of the role of opioid antagonist naltrexone in 10 studies each of ASD [194] and self-injurious behaviour [195] found that oral naltrexone was effective in reducing the symptomatology in 77% of 128 cases and in 50% of 124 cases respectively. | |
| Particularly intriguing results came from the genetic mouse studies mentioned [186,193]. The very high rate of clinical syndromes which include mental retardation and / or ASD amongst X-linked diseases is well known [196]. Mouse models of the Fragile-X syndrome and the MOR-KO disorders form the best models in which to study ASD [186]. It has been shown that both the MOR gene Oprm1 and the gene for CRH, Crh are under the control of MECP2 (methyl-CpG-binding protein 2) which is the key coding gene on the X-chromosome located at Xq28 duplications (and triplications) of which are associated with autism and anxiety related disorders [193]. MeCP2 binds to the promoter of the Opmr1 and in the context of copy number variants, increases MOR expression and accounts for the heightened anxiety behaviours and social distress. Contrariwise, pharmacological activation of the MOR was associated with increased sociability [193]. | |
| Such opiate-related immunogenic activities including alterations of neurogenic stem cell activities, dendritogenesis, synaptic and dendritic pruning, and neurite guidance and connectivity may also contribute to the elevated incidence of neuropathologies seen in adults exposed to immunogenic drugs of abuse, as alluded to above. | |
Section III: Central Inflammation in the Aetiopathogenesis of Metabolic Syndrome |
|
| A powerful body of work primarily from a tram at Albert Einstein College of Medicine led by DongshengCai shows that low grade hypothalamic inflammation is linked to various aspects of the metabolic syndrome including hypertension, obesity, glucose resistance, hyperglycaemia including diabetes, and dyslipidaemia [46,49,59,197-204]. This work is relevant to opiate dependence because various authors have described the changes of the metabolic syndrome occurring in opiate dependence [74,128]. Some of the landmark findings from this group can be summarized serially as follows. | |
| In 2008 it was shown that over nutrition activates an atypical level of inflammation in the MBH which underlies both glucose and leptin resistance [46]. Forced suppression of this inflammation in the whole brain, the MBH or the agouti-related protein neurons of the MBH suppressed the development of overweight and glucose intolerance. It is important to note that whole brain and MBH inflammation had similar effects. The effect of NF-κB was mediated through SOCS-3 (suppressor of cytokine signalling-3). This effect was shown to be mediated through endoplasmic reticulum (ER) stress. As noted below, since long chain fatty acids can bind to TLR4, which can be endocytosed into phagocytic vacuoles and thereby communicate with the ER system, such a system is eminently plausible to lie at the root of over nutrition related hypothalamic neuroinflammation. Indeed two of the three pathways mediating ER stress, PERK (Protein kinase RNA-like Endoplasmic Reticulum Kinase)and eIF2α (eukaryotic initiation factor 2α), were noted to be activated by the high fat diet (HFD). Importantly, ER stress was noted to be both a cause and an effect of NK-κB activation, such that NK-κB and ER stress feed forward to amplify each other [46]. One schematic drawing of NF-κB activation is shown in Figure 3 (Qiagen Inc.). | |
| Figure 3: NF-κB family pathway. | |
| These observations are of particular relevance to the biology of ageing for several reasons. Firstly endoplasmic reticulum stress has recently been shown to be linked to modulation of telomerase activity [205]. Secondly telomere length has in turn has been shown to be linked to epigenetic state by modulation of the Suv39h1 and Suc39h2 histone methyltransferases [206-208]. And finally ER stress is metabolically stressful for cells, and, when uncontrolled, can lead to cellular apoptosis by variety of mechanisms. | |
| In 2011 Zhang et al., performed an elegant study of the influence of hypothalamic neuropeptide secretion on resistance to obesity development under a high fat feeding paradigm [202]. They noted firstly that “hypothalamic neuropeptides play essential roles in regulating energy and body weight balance”, and particularly the balance between anorexigenic α-melanocyte stimulating hormone (α-MSH) and cocaine-amphetamine related transcript (CART) on the one hand and orexigenic neuropeptide-Y (NPY) and agoutirelated protein (AGRP) on the other. Whilst these neuropeptides are generally controlled at the level of the transcriptional factors which regulate their gene expression, they further showed that control of hypothalamic oxytocin release by negative regulation of synaptic release of oxytocin granules through synatpotagmin-4 could protect against an obesogenic environment. | |
| Autophagy is the process by which old cellular components are recycled by the cell for re-utilization into new cellular components under normal homeostatic conditions and particularly under conditions of cellular stress such as caloric restriction or organellar dysfunction. Pathways upstream of autophagy therefore include IKKβ (IκB Kinase) /NF-κB (Nuclear Factor-κB), TLR’s including TLR4 and MyD88 (myeloid differentiation factor 88), ER stress and JNK (Jun-N-terminal kinase) – AP1 (Activator Protein 1) signalling [201]. A further paper from this group in 2011 studying the key mediator of autophagy known as autophagy related protein 7 (Atg7) showed that the development of obesity was associated with an inhibition of hypothalamic autophagy [201]. Moreover the effects of Atg7 inhibition were mediated by central inflammation by IKKβ/NF-κB, and inhibiting hypothalamic NF-κB activation reversed this effect including the phenotype of the development of obesity and insulin- and leptin- resistance. Since autophagy inhibits inflammation [209], inhibition of autophagy is associated with a pro-inflammatory hypothalamic milieu [210-214]. Inhibition of the NLRP3 inflammasome activity is partly responsible for this antiinflammatory activity. Contrariwise stimulation of autophagy, as occurs in caloric restriction, is strongly anti-inflammatory. Other cellular events which are associated with IKKβ/NF-κB activation include ER stress, cellular oxidative stress, and mTOR (mammalian Target OfRapamycin) activation. Furthermore both ER stress and oxidative stress are associated with autophagy defects; and mTOR has been shown to be activated by elevated levels of plasma leptin [201]. | |
| Further work from this laboratory showed that induction of brain ER stress (with thapsigargin) was sufficient to cause glucose intolerance, systemic and hepatic insulin resistance, and blood pressure increase in as little as three days [48]. These changes were mediated by an increase in sympathetic tone, which was prevented by sympathetic blockade. Inhibitions of either ER stress (with tauroursodeoxycholic acid) or its downstream target NF-κB were effective in preventing these changes. Hence the sequences of events outlined were: obesogenic diet – hypothalamic ER stress - NF- κB activation – elevated sympathetic tone – peripheral metabolic syndrome. It was further noted that pharmacologic of genetic modulations of the hypothalamus could induce feeding and body weight changes without the alteration of diet or exercise parameters. ER stress was noted to be inducible by a variety of cellular stresses including protein misfolding, redox imbalance, energy consumption and calcium dysregulation. The last three items on this list have been noted in opiate dependence [74,129,130,215]. A schematic illustration of NF-κB activation by ER stress is shown in Figure 4. A fascinating study from the Cai laboratory in 2011 showed that hypothalamic glucose sensing was under the transcriptional control of POMC (proopioimelanocortin) which was in turn profoundly positively up-regulated by HIF ( Hypoxia Inducible Factor), a powerful transcription factor activated by environmental and pathological hypoxia which acts to induce metabolic adaptation, vascular growth and cellular survival [204]. HIF loss of function was associated with an impairment of the hypothalamic glucostat function and the development of obesity. These effects of HIF on POMC gene were independent of leptin and ACTH signalling. These obesogenic features of HIF deficiency could be rescued by hypothalamic lentiviral delivery of HIF gene into the hypothalamus. HIF regulation by hypoxia has been shown to be mediated by reactive oxygen intermediates regulating the leakiness of electrons from mitochondrial complex III. Normoxic regulation of HIF also occurs in response to activation of the PI3K (Phosphoinositol-3-Kinase) -mTOR cascade and the SIRT1 sirtuins (histone deaceytlases). This work showed that POMC is a key molecule involved in the hypothalamic glucostat and lipostat, and thus the POMC neurons are central to real time glucostatic regulation of feeding and metabolic regulation. The hypothalamus was also noted to be directly sensitive to circulating nutrients including glucose, amino acids and fatty acids. These reactions were mediated via the POMC breakdown product α-MSH. Many species from rodents to humans contain HIF response elements in the promoter of the POMC gene. Mutations of the POMC gene or its α-MSH signalling pathway (including their receptors particularly melanocortin receptors 3 and 4 (MCR3 and 4) ) were noted to cause obesity from rodents to humans. The HIF response element was noted to occur immediately upstream of the TATA-box POMC transcription start site (TSS). Overexpression of HIF1α and HIF 2α increased POMC reporter gene expression by 12- and 26- fold respectively. When HIF1α and HIF2α were overexpressed together with HIFβ the heterodimeric complex stimulated POMC gene expression by 362- and 466- fold respectively [204]. HIF2α was the isoform most expressed in the brain, and was abundant in the mediobasal hypothalamus and the neurons of the arcuate nucleus. The authors also noted that two response elements for STAT3 (signal transducer and activator of transcription 3) one of the primary immune-active transcription factors, and also the key mediator of leptin’s action, occurred upstream of the POMC TSS. HIF was shown to be crucially required for POMC glucostat signalling. Mediators of HIF sensing were the tricarboxylic intermediates fumarate and succinate, the cytoplasmic energy sensor adenosine monophosphate (AMP) kinase (AMPK), and the key anabolic activator mTOR-S6K (S6 kinase). HIF/POMC knock-out mice fed normal chow were hyperphagic, and although not obese had an (estimated) 85% increase in fat mass, together with an increase in the size of their fat cells. There was an impaired thermogenic response to re-feeding in brown fat. When these knock-out mice were fed an obesogenic diet they became obese much faster than the pair fed controls. Administration of complete HIF dimers in these situations rescued these metabolic phenotypes. | |
| Figure 4: NF-κB activation by viruses. | |
| A further remarkable paper from this group in 2011 showed that IKKβ/NF-κB – POMC activation was a critical mediator of the development of hypertension in mice exposed to an obesogenic environment, that MBH POMC neurons were thus central to the development of essential hypertension under this model, and that the effect developed rapidly within just a few hours of exposure to a prototypical inflammatory stimulus delivered by TNFα into the third ventricle [216]. Moreover the pro-hypertensive effects of the central TNFα delivery could be overcome by genetic ablation of IKKβ/NF-κB signalling in the MBH POMC neuronal population. This effect was not seen with similar genetic modification of AGRP/IKKβ knockout mice. The authors note that chronic inflammation in peripheral tissues is a recognized feature of both obesity and hypertension. Similar inflammation has also been documented in the hypothalamus [48]. By looking at low and high frequency blood pressure variation ratios and noradrenalin release, the authors were able to demonstrate increased sympathetic tone in mice exposed to constitutively active hypothalamic IKKβ as the basis for the hypertension which developed. Blockade of the MBH IKKβ was shown to prevent the hypertension induced by high fat diet feeding paradigms. Very interestingly these authors studied the effects of TNFα activation in the two main neuronal populations of the MBH. Basal expression of IKKβ / NF-κB was negligible in both groups of neurons, but after TNFα stimulation they found that while IKKβ phosphorylation (on tyrosine 188) was minimal in AGRP/NPY neurons, it was quite profound in the POMC/CART neurons. This was in part related to a much more pronounced expression of TNFR2 in the plasma membrane and cytoplasm of the POMC compared to the AGRP neurons. This suggests that the hypothalamic MBH POMC neurons are the primary target of hypothalamic inflammation and the prime culprits of obesity-related sympathetic hyperactivation and therefore cardiovascular - metabolic disease, much moreso than the AGRP/ NPY population. | |
| These authors penned a provocative review of this field in 2012 [48]. They noted that the term “metaflammation” or “metabolic inflammation” had been coined for the low grade inflammation characteristic of chronic diseases of the metabolic syndrome / syndrome X cluster (hypertension, hyperlipidaemia, hyperglycaemia, insulin resistance) and its accompanying complications including type 2 diabetes, coronary artery disease, stroke, atherosclerosis fatty liver disease and ageing related degenerative diseases. They note that activator protein 1 (AP1) is also involved in the genesis of these changes at the molecular level, and further that TLR’s 1, 2, 4 and 6 had been implicated in the binding of fatty acids to cellular membranes and the induction of inflammation. Three pathways were described for the induction of over nutrition related hypothalamic inflammation including nutrient transporters, the toll-like receptor pathways, and the cytokines / chemokines pathways. Mitochondria were noted to be very sensitive to increased oxidative stress including the overabundance of reactive oxygen / nitrogen species (ROS / RNS), and increased electron leakage was noted from dysfunctional stressed mitochondria, which in turn resulted in increased oxyradical flux. Moreover both inflammatory states and obesity are associated with increased oxyradical formation, and brain neurons are highly susceptible to free radical stress and moreso under inflammatory conditions. Furthermore mitochondrial ROS have been linked by several studies to activation of the NLRP3 inflammasome, a powerful multimolecular platform for generation of activated interleukin-1 (IL-1) and interleukin-18 (IL-18) [217-220]. The three major canonical pathways of ER stress and the unfolded protein response were activating factor 6 (Atg6), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and downstream eukaryotic translation initiation factor 2α (eIF2α), and inositol requiring kinase 1 (IRE1) and these were all noted to be in the induction and maintenance of hypothalamic metabolic inflammation. The obesity-related autophagy defect was again noted to be pro-inflammatory [201]. They note that cellular stress originating in mitochondrial oxidative stress and ER stress can spread from one organelle to another via the highly interconnected intracellular network of ER membranes leading to central neuronal inflammation and peripheral metabolic dysregulation. Toll-like receptors (TLR’s) have been shown to be stimulated by lipids to induce proinflammatory changes in adipocytes, myocytes and macrophages. Central TLR4 activation (in this case by oversupply of saturated fatty acids or high fat diets) was associated with weight gain and systemic insulin and leptin resistance, changes which were blocked with whole body or brain TLR4 abrogation. TLR4 activation was also known to be associated with ER stress (consistent with the pathway of TLR-ligand endocytosis and internalization via an endosomal-phagolysosome pathway) and neuronal apoptosis (particularly in association with TNFR activation. Sirtuin activation was noted to be anti-inflammatory. Hypothalamic signalling countering hypoglycaemic events is mediated in part by GABA and glutamate signalling all of which are deranged in central inflammatory states. Altered POMC neuronal electrophysiology has been noted in obesity. These changes identify hypothalamic inflammation as a central therapeutic target for a range of metabolic disorders related to over nutrition, chronic central inflammation, and central TLR activation. All of these key ingredients will be noted to occur to some extent in opiate dependency. | |
| Another interesting review in 2011, notes that TNFα is both a stimulus of IKKβ/NF-κB and a target of its gene transcription [197]. Similar remarks apply to IL-1, IL-6 and AP-1 [199]. Clearly this establishes several powerful proinflammatory positive feedback loops driving such inflammation. | |
| A further review in 2012 noted that over nutrition causes an atypical form of inflammation triggered by the innate immune system leading to metabolic derangements at the cellular, organ and systemic levels [199]. This metabolic inflammation is linked to the induction of various intracellular stresses such as mitochondrial dysfunction, ER stress, and an autophagy defect. Moreover these processes occur also in the hypothalamus, which, since it controls metabolism, appetite, energy expenditure, carbohydrate and lipid metabolism, blood pressure homeostasis and the tone of the sympathoadrenal autonomic nervous system, can all become deranged. ROS are noted to be produced in inflammatory states from numerous sources including the leaky electron transport chain, NADPH oxidase, cyclooxygenases, lipooxygenases, xanthine oxidase and cytochrome P450 enzymes. Pathological accumulation of such ROS is associated with diverse diseases including metabolic syndrome, cardiovascular diseases, type 2 diabetes (T2DM), neurodegenerative diseases, brain ageing and cancer – all of which have been noted at increased frequency in opiate dependence. Moreover mitochondrial dysfunction in MBH POMC neurons has been linked with impairment of central glucose sensing. ER stress can trigger either cellular apoptosis or cellular inflammatory pathways, and has been linked with obesity, insulin and leptin resistance, T2DM, cardiovascular disease, cancer and neurodegeneration. Moreover brain ER stress has now been shown to be a central and pivotal organizer of these changes at the systemic level. | |
| The primacy of brain TLR4 signalling was noted to have been demonstrated by a study which showed that brain specific deletion of TLR4 prevented central leptin and peripheral insulin resistance and weight gain under a high fat feeding regime [221]. | |
| Cytokines can access the brain through circumventricular organs (organumvasculosum of the lamina terminalis, the sub-fornical organ, the median eminence of the hypothalamus and the area postrema) choroid plexus and leptominenges [183], and also act in paracrine manner as cellular inflammation spreads to nearby cells in the case of most of the cytokines. NF-κB is itself known to be a redox sensitive transcription factor, particularly by ROS-induced alternative phosphorylation of IκBα which releases NF-κB from its inhibition, and by oxidation of sensitive sites in NF-κB pathway phosphatases including IKK’s (α,β,γ) and PTEN (Phosphatase And Tensin Homologue) and their downstream targets including Akt (protein kinase B). NF-κB activation then induces the release of many more strongly oxidating species. gp130 signalling (via LIF (Leukaemia Inhibitory Factor)) to JAK1-STAT3 is also redox sensitive [222]. It is likely that brain oxidative stress arises directly from over nutrition which drives brain mitochondria harder and thus increases their electron leak, and through activation of brain and hypothalamic NF-κB. TLR4 activation was shown to lead to NF-κB activation through both early MyD88 –dependent and late MyD88 –independent pathways, and to induce ER stress. The effects of hypothalamic IKKβ/NF-κB activation were dependent on the cell type involved, for in the AGRP/NPY neurons it induced energy imbalance and obesity, whereas in POMC/CART neurons it induced hypertension and glucose intolerance. Physiological hypothalamic nutrient sensing by leptin occurs via the JAK2 (Janus Activated kinase) – STAT3 pathway and insulin signalling via the PI3K/Akt pathway, however both are disrupted under pathological conditions by proinflammatory signalling mediated importantly via IKKβ/NF-κB. Importantly anti-inflammatory treatment with either aspirin or the cannabinoid Receptor 1 (CB1) antagonist rimonabant or numerous other inhibitors of these pathways have been shown to be associated with weight loss or improvement of other metabolic or pathological parameters in overweight and obese humans. Exercise was noted as having its anti-inflammatory effect by stimulating the anti-inflammatory cytokines IL-4 and IL-6. | |
| An important paper from the Cai group in 2012 showed that high fat diet (HFD) feeding of adult mice led to a profound inhibition of the normal neurogenic renewal processes of the mediobasal hypothalamus, in a manner mediated by IKKβ/NF-κB induced apoptosis and downstream notch signalling [49]. Reversal of IKKβ/NF-κB signalling or blockade of the notch pathway was able to dissociate the effects of HFD and neurogenic impairment. MBH POMC signalling is known to be impaired by HFD, but loss of POMC neurons had also been recently identified, related in part to a neurogenic defect. The present study demonstrated markedly impaired POMC neuronal survival, proliferation and differentiation. Indeed the neurogenic potential of the normal MBH was significant, and comparable to that occurring in the dentate gyrus of the hippocampus, with the former being about half the latter. The number and size of neurospheres cultured from HFD MBH were both fewer (about half) and smaller than those from pair-fed controls. Moreover the impairment of neurogenesis in the MBH (about 50%) was much more marked than that which was seen in the well-recognized neurogenic subventricular and subgranular zones (about 20%). Importantly it was shown that the reason for the impaired growth of the cultured neurospheres was the paracrine release of the cytokines TNFα and IL-1 via IKKβ/NF-κB. Blockade of these positive feedback loops restored the neurogenic potential of the cultured neurospheres. Similarly blockade of MBH microglial IKKβ under HFD in situ also restored the neurogenic defect. The rate of apoptosis of MBH neural stem cells was shown to be increased 9-fold, a finding related to the fact that the apoptotic genes Bim, Bax, Bnip3 and caspase-3 were all target genes of NF-κB. Notch isoforms 1 and 4 and delta-like ligand 4 were all found to have NF-κB response elements in their promoters, to be upregulated under NF-κB stimulation and HFD, and that their inhibition led to a restoration of neurogenic potential. In mice injected intracerebroventricularly with constitutively active IKKβ and followed for three months the number of hypothalamic neural stem cells (NSC’s) reduced 60%, resulting in a 10% reduction in total POMC neurons after that period, but a negligible effect on NPY neurons. After this period the mice demonstrated hyperinsulinaemia, glucose intolerance, over eating and weight gain. At 10 months post-gene delivery they were severely obese despite being maintained on a normal chow diet. These data show that the adult generation of new NSC’s in the MBH is critically important for the prevention of severe systemic disease. | |
| In a review in 2012 Cai noted that IL-6, as produced in obesity and T2DM, was associated with the induction of degeneration of forebrain GABAergic neurons [198], subsequent to NF-κB activation and the NAPDH oxidase-dependent generation of superoxide [223]. GABAergic neurons are essential for normal information processing, encoding and retrieval in the hippocampus and cerebral cortex. Moreover potentiation of brain oxidative stress in diseases as diverse as Huntingdon’s disease, Parkinson’s disease, and diabetes were associated with significantly worse neuronal outcomes. | |
| Building on this stellar research portfolio the Cai laboratory recently published an amazing tour de force examining the role of the hypothalamic inflammation now well described by them in its interaction with the determination of longevity [59]. | |
| This work produced the following remarkable results: firstly that the hypothalamus coordinates and controls lifespan determination; that neuronal-microglial IKKβ/NF-κB based cross-talk was central to this role; and that much of this effect was attributable to the IKKβ/ NF-κB induced suppression of GnRH (Gonadotrophin Releasing Hormone). Because the effect of the latter was identical in both sexes, this action of GnRH was not thought to be related to its secondary actions on gonadal sex steroids. GnRH supplementation repaired the age-related neurogenic defect, and decelerates many age-related biomarker declines. Hence both MBH immune suppression orGnRH were noted to be candidates for anti-ageing therapies. This study is described in further detail in Section V. | |
| The hypothalamus has thus been shown to be a key neuroendocrine area integrating hormonal, neural, nutritional and immune inputs and coordinating metabolic regulation, thermogenesis, circadian rhythms, endocrine orchestration and the organismal ageing mechanism . POMC neurons play a key role in integrating most of these functions, and in particular signal satiety, and tend to reduce glycaemia, lipaemia and ameliorate ageing processes systemically. These neurons are suppressed by exogenous opiates. The case is compelling therefore that exogenous opiate induced hypothalamic inflammation is causally related to the induction of disease and systemic ageing processes. | |
Section IV: Central Opiate-Induced Neuroinflammation |
|
| A large and well established literature now shows that opiates and most other addictive substances are associated with central neuroinflammation. In the case of the opiate group of drugs, this occurs because the central opiate morphine nucleus fits like a key into the binding pocket of MD2 (Myeloid Differentiation Factor 2) one of the binding partners of TLR4 [224-228] (Figure 5 from Hutchinson 2009). This triggers the complex and powerful and phylogenetically highly conserved TLR4 endotoxin receptor signalling mechanism (see Figure 6, from Hutchinson 2011). Evidence has been presented implicating TLR’s 2, 4, 7 and 9 in opiate pharmacology [25,93,224,229-234], with most of the work centring on the role of TLR4. Particularly in microglia TLR4 ligation triggers the powerful arsenal of immune active molecules as summarized in Figure 7 [235]. In terms of understanding the pathophysiology of opiate dependence it is hardly possible to overstate the importance of TLR4 signalling (Figure 8). However the profound implications of long term low level stimulation of this conserved and foundational immunobiological signalling pathway have hardly begun to be appreciated in clinical addiction practice. | |
| Figure 5: In Silico computerized modelling of opioid-TLR4-MD2 binding pocket Watkins (2009) “The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia” Trans in Pharmacological Sciences, 30 (11), 581-591 Used by Permission. | |
| Figure 6: Simplified schema of main downstream intrinsic and extrinsic pathways from TLR signalling Hutchinson (2011), “Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia” Pharmacological Review, 63 (3), 772-810. Used by Permission. | |
| Figure 7: Main domains and receptor-ligand pairs of macrophage signalling. From Lucin (2009), Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64 (1): 110-122. Used by Permission. |
|
| Figure 8: Main pathways of TLR family signalling. | |
| A partial listing of the effector pathways of this system may be made from the explicit comments in Figure 6 and their downstream implications and elsewhere [183,236,237], and listed as: | |
| Ceramide and Sphingosine Signalling [238], | |
| NADPH Oxidase activation (ROS released); | |
| TLR4 Lipid Rafts | |
| PI3K / Akt Signalling, Apoptosis | |
| NF-κB signalling | |
| Interferon’s, Types I and II | |
| MAP Kinase pathway activation (JNK, ERK and P38) [239] | |
| AP1 Activation (c-Jun and c-Fos) | |
| Prostaglandin (Cyclooxygenase) / Lipooxygenase activation | |
| Heat Shock Protein Intracellular Stress System Activation (70kD and 90kD) [25,93,183,236,240] | |
| Endoplasmic Reticulum Stress (Implied by (4 & 10) | |
| Calcitonin Gene Related Peptide (CGRP) | |
| Neuronal, Inducible and Endothelial Nitric Oxide Synthase (nNOS, iNOS and eNOS) [241] | |
| Cytokines and Chemokines, IL-1β, IL-6, TNFα, CCL2, CX3CL1, CXCL12, CCL3, CCL5, CCL20, IL-10, IL-4, IL-12 | |
| Caspase 3 [236] | |
| Elevated Glutamate (and aspartate) levels, glutamate transporter dysfunction, and key protein nitration / oxidation, and heightened NMDA and AMPA receptor excitability [183], | |
| TGFβ stimulation | |
| Matrix metalloproteinase activation ((MMP-2, MMP-4, MMP-9) | |
| Purinergic signalling via P2X4, P2X7, P2Y12 [183,237] and others | |
| STAT3 expression | |
| JAK2 activation | |
| Protein Kinase C expression | |
| cAMP response element activation | |
| Calmodulin protein kinase II activation (CaMKII) | |
| Alteration of neurotrophic factor release such as Glial Derived Neurotrophic Factor (GDNF) [183,242] | |
| NLRP3 inflammasome activation [183] | |
| ROS induction (superoxide and nitric oxide etc.) [183,236] | |
| NOD2 stimulation (morphine action via TLR2) [231] | |
| Epigenetic and Chromatin Remodelling [243] | |
| Impaired Neurogenesis [244] | |
| Neuronal Apoptosis particularly affecting POMC neurons [183,239] | |
| The downstream signalling from TLR4 activation by opiates is complex. One of the most comprehensive recent reviews of this subject is that prepared by the Adelaide-Colorado-Maryland group in 2011 led by pioneers in this field, Linda Watkins, Mark Hutchinson, and Kenner Rice [25]. This review and their associated voluminous corpus of work describe the complexities of the downstream TLR4 signalling in considerable detail. Their 2011 review is stratified by brain area, by cell type and by drug type. Mechanistically it is summarized in Table 2. It is not necessary to elaborate on these complex nuances at length, as it is well handled in the many papers from this group. The hypothalamus has been specifically implicated as being a field prominently involved in many of these observations [25]. | |
| It should be noted that the neuroimmune changes noted in this body of work complement rather than supercede the role of derangements and perturbations in mesolimbic system dopamine, monoaminergic and glutamatergic signalling as has already been well described elsewhere [24,25,242]. | |
| In the context of this impressive body of evidence for immunostimulation it appears at first somewhat paradoxical that opiate and other drug dependencies have long been associated with an increased incidence of viral and other infections, and much clinical evidence has been presented that opiate dependent patients are immunosuppressed [33,79,245-250]. This apparently paradoxical situation has been addressed by thoughtful authors who have noted that evidence for both immune stimulation and immune suppression exist, with both being different expressions of the immune stimulation and dysfunction [25,33,93,247], much as has been noted with clinically available immunosuppressive drugs cyclosporin and fingolimod [251-254], and indeed as is well recognized pathologically in the common clinical rheumatological disorders rheumatoid arthritis and systemic lupus erythematosus [4,5]. | |
| It should also be noted that whilst for purposes of analysis receptors and their downstream pathways are typically analysed together, in fact they can co-locate and directly influence each other’s signalling pathways, and commonly share similar intracellular transduction systems [25]. Hence CXCR4 can profoundly modify TLR4 and TLR2 signalling, and most of the TLR’s share common intracellular transduction pathways with IL-1 (hence the designation the Toll/IL-1 Receptor (TIR) intracellular transduction signalling cascade). Moreover there is signficant overlap between TLR4 binding and binding to intracellular scaffolding proteins such as filamin A [25]. | |
| Whilst the receptors of the innate immune system such as the TLR’s are commonly said to respond to PAMP’s (pathogen associated molecular patterns) and DAMP’s (damage associated molecular patterns), their ligation of the opiate class of agents and other addictive chemical classes has opened up discussion of their association also with XAMP’s (xenobiotic associated molecular patterns) [242,255]. These authors repeatedly make the point that whilst uptake of exogenous opiates by classical opiate receptors is well described, only 2% of the administered opiates is taken up by classical stereospecific binding sites, with the remainder being taken up by non-stereospecific sites [256] including the TLR’s [25,93,224,225,228,257]. | |
| Importantly, as shown in Figure 9, after Hutchinson 2009, the opiate antagonists +-naltrexone and +-naloxone, have no activity at classical opiate receptors, yet powerfully reverse these neuroimmune stimulatory actions of opiates [257]. This clearly implies that their actions in this respect are not occurring at sites of classical opiate receptor binding. Moreover it has also been shown that in some situations naltrexone can act as an inverse agonist [258]. | |
| Figure 9: In Silico MD2 modelled binding compared to In Vitro TLR4 signalling, tested in (a) the absence and in (B) the presence of ibudilast (AV411) Watkins (2009) “The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia” Trends in Biological Sciencess 30 (11), 581-591 Used by Permission. | |
| It should be noted that the pro-inflammatory environment is highly oxidizing, as most of the products involved either themselves are actively oxidizing, or frequently give risk to such species. An increased rate of inflammatory disease has also been noted in drug dependent patients [25,93]. This is important to the inflammatory theory of ageing which notes that increased ROS are a key correlate of the ageing process [259-263]. Indeed heightened inflammation is one of the major theories of the induction of ageing [4,120-124,264-266]. | |
| The major point to emerge from this is that chronic opiate administration is associated with cerebral and mediobasal hypothalamic inflammation evident mainly at the molecular level, just as has been described for HFD- and obesity- related hypothalamic inflammation examined in detail by the work of the Cai laboratory. | |
| Some readers may wish to familiarize themselves with studies which preceded those of the Cai laboratory and formed the conceptual underpinnings for much of its far reaching insights. To review these studies in any detail is beyond the remit of this review but a brief summary with recommended additional reading is as follows. | |
| TLR4 physiology, downstream signalling pathways | |
| Since TLR4 is clearly at the centre of the new conception of molecular pathophysiology of these related syndromes, it is appropriate to consider its upstream triggering mechanism with particular reference to metabolic and immune stressors and its downstream signalling pathways in further detail, and in so doing to review briefly the studies which preceded that of the Cai laboratory and formed the conceptual underpinning of its far reaching and penetrating insights. Reference is again made to Figures 3-8. | |
| LPS (Lipopolysaccharide) binding to TLR4 also requires LBP (LPS Binding Protein) CD14 and MD2 [267]. MD-2/TLR4 then dimerizes to transmit the activation signal intracellularly [267]. Upon ligand binding the TLR4/MD2 complex is internalized into endophagolysozomes where it delivers its immune activating signal [268]. In the ER MD-2 is phosphorylated on two tyrosine residues by Lyn kinase (one of the src stress related kinases) [267]. This implies that trafficking occurs from the ER and Golgi apparatus where TLR4 is synthesized to the cell membrane and back down again after receptor ligation and binding. Insufficiency of ER nascent TLR4, or its cochaperone molecules GRP94 (Glucose Regulated Protein 94) and GRP78 can overextend this system and induce ER stress [269]. TLR4 can also signal from its intracellular location within the ER [267-268]. Interestingly TLR4 signals to NF-κB; however TLR4 gene is an NF- κB target so that TLR4 induces its own synthesis [270]. GRP78 is a member of the Hsp70 family [271]. | |
| TLR4 priming can occur. Long chain unsaturated fatty acids such as palmitate and stearate are toxic to endothelial cells, fibroblasts, pancreatic β-cells, hepatocytes, and myoblasts, and can trigger ER stress. The serum levels of long chain fatty acids are elevated in obese individuals. After preparation by exposure to the TLR4 ligand palmitate, exposure to LPS can induce cell death after TLR4 binding and internalization by TRIF-dependent activation of intracellular lysozomes [272]. Similarly cross-talk between TLR4/MyD88/NF-κB and scavenger receptors mediates macrophage foam cell formation [273]. | |
| Whilst it had been known for some time that saturated fatty acids (SFA’s) produce an inflammatory response in the hypothalamus, the exact biochemical pathway by which this occurred was unclear. It was shown that SFA’s bind to TLR4/MD-2 and are internalized into the ER system and trigger NF-κB [50]. Moreover this process induces leptin- and insulin- resistance [50]. ER stress induces the unfolded protein response (UPR) which can also activate NF-κB [50]. Cytokines had been known to induce insulin resistance in peripheral tissues and this work demonstrated that cytokines arising either as a result of TLR4 ligation or ER stress could also trigger similar changes in the hypothalamus [50]. In this work the HFD was associated with elevated hypothalamic levels of IL-1β, IL-6 and TNFα, central insulin- and leptin- resistance and body weight gain [50]. These changes were mediated mainly through microglia rather than other cell types. Other work showed that SFA priming of dendritic cells in peripheral adipose tissue was dependent on TLR4 signalling and NLRP3 inflammasome formation and induction of insulin resistance [274,275]. | |
| When TLR4 is engaged after ER stress induction in atheromatous plaques, the combined effect of scavenger receptor and TLR4 activation can result in macrophage cell death [276]. | |
| It is not surprising therefore those high fat diets have been associated with the induction of hypothalamic damage detectable both histologically in adipogenic model organisms, and by MRI in obese humans [66]. Whereas peripheral inflammation was noted to develop over weeks to months, in the hypothalamus an early inflammatory change and NF-κB activation was noted within the first 24 hours. Markers of arcuate neuronal inflammation were seen after one week of HFD in association with reactive gliosis and recruitment of microglia and astrocytes [66]. Under HFD microglia assumed an enlarged and activated morphology. The reactive gliosis was marked. Astrocytes formed what appeared to be a syncytium as their processes intermingled and interwove. Whilst this effect was initially transient and resolved within 2-3 weeks, it re-appeared in mice fed HDF for 8 months [66]. Elevated levels of Hsp (heat shock protein) 72 were identified in the hypothalamus, particularly in POMC neurons. Indeed after 20 weeks of HFD feeding 81% of POMC neurons contained autophagosomes compared to only 8% of mice fed control laboratory chow (P<0.0001). These POMC neurons also contained abnormal mitochondria which were swollen, had irregular cristae, had short and discontinuous cristae and were fewer electrons dense [66]. Moreover these stigmata of intracellular injury were accompanied by a dramatic reduction of POMC cell numbers by 25% after 8 months HFD exposure [66]. The authors noted that the POMC cells play a critical role in the protection against obesity, and that a reduction in their numbers alone is a sufficient cause of excessive weight gain. | |
| Similarly systemic administration of LPS was noted to induce ultra structural changes of ER stress and dilatation, autophagy, mitochondrial stress and dilatation, and necrosis and molecular evidence of cyclooxygenase, lipooxygenase, iNOS and TLR4 mRNA induction a few hours later [277]. This is consistent with the above schema of TLR4-induced TLR4 transcription [270]. | |
| It is relevant to this review that increased rates of release of free radicals during the neutrophil oxidative burst have been noted in hypertensive patients [278], and moreover that renal artery denervation via catheters has been shown to reduce blood pressure in clinical series, by way of reducing the increased input of central sympathetic efferent tone to key renal structures including the glomeruli, renal tubules and juxtaglomerular apparatus [279]. Moreover in mammals TLR4 activation has been shown to contribute to blood pressure elevation [270,280]. Importantly TLR4 has been noted to be activated both by angiotensin II and by C-Reactive Protein (CRP) [270]. Dramatic elevation of CRP has been noted in clinical opiate dependence [16,17]. TLR4 is also responsible for elevated levels of Hsp60 and Hsp70 seen in essential hypertension [270]. Moreover the elevated levels of COX2 are responsible for the elevated levels of vasoconstrictor prostanoids which are thought to account for some of the increased vasomotor tone seen in hypertension [270]. | |
| The opiate antagonist naltrexone was shown to protect against a model of septic shock by an indirect mechanism consistent with TLR4 antagonism [281] as has been described elsewhere [25,225,257]. | |
| TLR4, ERStress and AutophagicDefects | |
| As noted earlier the inflammatory response is strongly oxidizing. Inflammation is largely coordinated by NF-κB. However NF-κB is itself redox sensitive. This suggests that there is a bidirectional effect of oxidizing conditions on inflammation. It has been shown that the UPR and ER stress are centrally involved in mediating this [282]. | |
| For the reasons mentioned at the outset the TLR’s and NLR’s stand at the intersection of immune activation and autophagy [283]. This has particularly been demonstrated in inflammatory bowel disease [210,283,284], and now also in the hypothalamus [46-49,198,199]. | |
| Given the co-occurrence clinically of dual opiate-benzodiazepine dependence it is also fascinating that one of the key autophagy chaperones is the GABA receptor-associated protein (GABARAP) which along with autophagy specific gene 8 (atg8) and microtubule associated protein light chain 3 (LC3) specifically direct phagocytosed cargoes to lysozomes, and induce lysozome dual membrane fusion [285]. | |
| TLR’s partners in autophagy induction include MyD88, TRIF, A20, TRAF-6 and Beclin-1 [286,287]. | |
| As noted elsewhere autophagy is strongly anti-inflammatory, probably by inhibiting P38 MAP kinase transcription [210]. | |
| As noted above ER stress can be induced by TLR signalling when the requirement for TLR trafficking outstrips the supply of cochaperone molecules [269]. Since its chaperones are induced much more slowly than TLR4 itself and at a much lower level by a factor of ten-fold, this can clearly occur [269]. This scenario is particularly likely upon prolonged stimulation of TLR signalling. | |
| IRAK2 (interleukin-1 receptor associated kinase-2) is a critical mediator of ER stress signalling [271]. This is important because IRAK2 is an important part of the IL-1/TLR (TIR) signalling platform. This in turn suggests that IRAK2 as well as TLR’s form a key point of intersection between innate immunity and ER stress pathways. ER stress was noted to have been identified as an important pathophysiological process in diseases as diverse as heart disease, diabetes, cancer, neurodegenerative disorders and inflammation. If the UPR is unsuccessful in restoring protein homeostasis the UPR can move the cell towards cell death by induction of CHOP (CCAAT/-enhancer binding protein (C/EBP) homologous protein) , stress associated kinases and the apoptotic machinery including Bcl-2 [271]. | |
| TLR4 knock-out was shown to be protective for ER stress induced by a HFD [288]. | |
| Oxidized LDL also induces macrophage ER stress via TLR4 [289]. | |
| POMC neurons not only contribute to, but are also targets of accelerated aging | |
| Fascinating recent reports demonstrated that, not only are hypothalamic POMC neurons key regulators of systemic metabolic and feeding processes, but they themselves are also subject to similar changes [290,291]. POMC neurons are often described as being larger than surrounding neurons in the arcuate nucleus, and this size enhancement can be particularly marked during aging. Cell growth is well known to be regulated largely by the mammalian target of rapamycin (mTOR), and the level of this key nodal regulator has been noted to rise within POMC cells with age [291]. Increased potassium influx hyperpolarizes and silences these cells, accounting for age associated obesity, and its pathological sequalae including hyperphagic obesity, cardiovascular disease, type 2 diabetes, neurodegenerative disease and cancer [290]. Obesity and hyperphagia were reversible on rapamycin or genetic mTOR inhibition [291]. Similar processes and silencing occurs in POMC cells subjected to enhanced opiate tone, both directly via MOR autoreceptors, and also by enhanced potassium flux through MOR-linked GIRK channels. This implies that the POMC neurons are not only major contributors to the dysmetabolic syndrome associated with age and opiate dependence, but are also themselves major targets of such processes. These relationships clearly establish a positive self-amplifying feed-forward loop whereby POMC neuronal age and dysmetabolic deleterious changes are amplified throughout the body, further stressing and aging the POMC-gated appetitive system. The large perikarya size of POMC neurons is thus evidence of these changes. | |
Section V: Lifespan Determination by Hypothalamic Neuroinflammation |
|
| It has long been established that perturbations of particular neurons in Drosophila and Caenorrhabditiselegans is associated with lifespan reduction or extension [60-64,200]. This work has recently been extended by a powerful genetic mutational study in mice by the Cai group (described earlier) showing that in addition to controlling much of the neuroendocrine regulation of the body, the hypothalamus exerts coordinate control of ageing [59]. This occurred through microglial originated inflammation which spread with age paracrine fashion to neighbouring cells, until in advanced old age over 90% of all the neurons of the mediobasal hypothalamus (MBH) stained positive for the activated subunit of NF-κB, known as phospho-Rel-A. Microglial NF-κB activation was also shown directly by insertion of a green fluorescent protein reporter construct prior to a microglial marker (CD11b). The intermediate early genes c-Jun and c-Fos were also shown to be stimulated by NF-κB activation. This finding has also been confirmed in other cell types [292]. These central changes were correlated with various peripheral stigmata of ageing including muscle fibre size, grip strength, cognitive ability, subcutaneous thickness, bone mass, and tail collagen crosslinking. | |
| Activation of NF-κB, c-Jun, c-Fos and protein kinase C were all shown to suppress gonadotrophin releasing hormone (GnRH). Age dependent GnRH release was then shown to be related to many of the stigmata of chronic ageing, and GnRH supplementation was shown to improve muscle size and endurance, dermal thickness, and cognition in Morris water maze testing. Interestingly the effects of GnRH infusion into the third ventricle were seen throughout the brain. Furthermore peripheral subcutaneous infusion of GnRH was also effective. The effects of GnRH were not thought to be acting via modulation of sex steroids, as they were closely parallel in both sexes. | |
| These results therefore lead to the demonstration that microglial mediated inflammation spreads to surrounding neurons with age, and thence throughout the brain, and that the systemic effects of this hypothalamic inflammation were spread throughout the body and brain partly by the suppression of GnRH. The authors also noted that it had been reported that NF-κB activation can also occur with age due to epigenetic modification at the NF-κB promoter [59]. Thus lifespan determination is linked to hypothalamic inflammation, metabolic dysregulation, and GnRH suppression, and the MBH is the central integrating and coordinating centre of this process organism wide. | |
| Section VI: Gut Microbiome and Hepatic Interactions | |
| The gut plays host to an enormous number of intestinal microbes, estimated at about 100 trillion. How this microbiota interacts with the host has recently become an area of increasing interest and formal study in disorders as diverse as obesity, metabolic syndrome, inflammatory bowel disease, bowel cancer and type I diabetes [77,293]. | |
| It is a classical observation dating from 1954 that experimental cirrhosis induced by a CCl4 or a choline deficient diet could be almost completely prevented by the administration of non-absorbable antibiotics [294,295]. | |
| Cells lining the bowel are subjected to a very high dose of gut microbe trafficking, but the enterocytes are relatively insensitive to such stimulation. It has been shown that shown that this insensitivity is related to the down-regulation of the TLR4 gene in intestinal enterocytes due to methylation of the TLR4 gene promoter [296]. Moreover this methylation state is dependent on the bacterial load of the large bowel, as it is much increased in germ free animals [296]. This indicates that epigenetic modification of important genes can occur related to the enteric bacterial load. It has also been shown that drugs [297-300], diet, pollution, and infections can also influence epigenetic state [296]. The bacterial product and short chain fatty acid butyrate has also been shown to act as an inhibitor of histone deacetylases, and therefore is an epigenetic modifier [296]. | |
| TLR4 engagement in gram negative sepsis has been shown to be an important trigger of Kupffer cell inflammasome activation and severe hepatic necrosis or death [301]. | |
| Hepatic fibrosis is a process known to be induced by PDGF (platelet derived growth factor) and TGFβ acting upon Kupffer and stellate cells after a variety of insults [302]. Hepatic injury is associated with increased levels of endotoxin in the portal and systemic circulations due to increased permeability of the enteric circulation. Whilst naive Kupffer cells are relatively inert to LPS, activated Kupffer cells are very sensitive to it at nanomolar levels [302]. It was recently shown that hepatic fibrosis is dependent on TLR4/MyD88/ NF-κB triggered down regulation of the TGFβ pseudoreceptor Bambi which allows TGFβ-induced fibrogenesis to proceed unhindered in a manner dependent on the resident gut flora and signalling from Kupffer cells to stellate cells [302]. Thus TLR4 signalling is the key molecular link between hepatic proinflammatory and profibrogenic processes, both of which are known to promote hepatocarcinogenesis [302]. LPS mediated TLR4 activation was thus shown to be central to the excessive fibrogenic wound healing responses seen in human cirrhosis. | |
| A recent analysis of the faecal microbiota of humans found that its diversity increased with advancing age, but was reduced with increasing frailty, comorbidity, reduced nutrition and markers of inflammation (CRP, TNFα, IL-6, IL-8) [293]. Hence a link between diet, frailty and increased ageing was demonstrated [293]. The suggestion was also made that these intestinal changes were also linked to the prognostically very important condition of immunosenescence of ageing [120-123,264,265,303-309], characterized by increased NF- κB activation and reduced numbers of naive T-cells [293]. | |
| MD-2 and TLR4 deficiency were shown to protect against nonalcoholic steatohepatitis in mice [310]. | |
| TLR4 signalling and downstream NADPH oxidase activation has been shown to be central to alcohol induced liver injury [76]. Moreover obese animals are more sensitive to endotoxin and develop steatohepatitis after lower doses of LPS, an effect which is believed to be related the gut microflora [76]. Hepatic stellate cells can respond directly to LPS, and can also be primed by Kupffer cell activation [76]. Elevated levels of TNFα and IL-6 after partial hepatectomy are believed to provide the initial stimulus for hepatocellular regeneration [76]. | |
| Weight gain, insulin resistance, elevated levels of blood pressure and serum lipids were noted to be transferable from normally colonized mice to germ free mice in a study examining the effect of conditional TLR5 knock-out [77,311]. These findings showed that gut microbiota contribute to metabolic disease, and implicate innate immune system malfunction in the pathogenesis of the metabolic syndrome [311]. Free fatty acids including palmitate were noted to stimulate TLR4 and also induce endoplasmic reticulum stress [77]. Obese humans have been shown to have different intestinal flora than non-obese humans [312]. This has been linked to the increased inflammatory state in obesity [312]. This was the source of the provocative title in one commentary written on this subject (in the journal Science) “The microbes made me eat it” [312]. | |
| Deterioration in intestinal transit time is also a feature of ageing [293]. | |
| Intestinal bacteria have been shown to be involved in riboflavin biosynthesis and carbohydrate metabolism [313]. The intestinal microbiome was felt to be relevant to human culture, nutrition, physiological variations and the impact of westernization in developing nations [313]. | |
| NOD2 (Nucleotide Oligomerization Domain 2) was the first susceptibility gene discovered for Crohn’s disease [314,315]. GWAS screens have since identified other innate immune system proteins including NOD1, IL-18R, IL-1RL1, CCL2, MUC19, CARD9, ATG16 and NLRP3 [314,315]. Many of these proteins interact functionally with NOD2. CD4 Th17 intestinal T-cell development is regulated by NOD2 and IL-23 pathways. ATG16L1 is a key regulator and recruitment protein involved in the unfolded protein response and activation of autophagy directed against bacterial invasion [314,315] so-called xenophagy [316]. Colonic colonization after birth was shown to be important for the development of isolated lymphoid follicles and the secretion of IgA by plasma cells [315]. NOD1 activation in the intestinal lamina propria has also been shown to enhance systemic innate immunity for gram positive and negative antigens [315]. Submucosal NLRP3 activity has also been shown to modify intestinal microflora. NOD2 also regulates microbial migration through colonic Peyer’s patches [315]. Both TLR’s and NLR’s (NODlike Receptors) have been linked with gastrointestinal carcinogenesis both in the stomach after Helicobacter pylori infection, and in the large bowel after colitis [315]. | |
| The incidence of Non-Alcoholic Fatty Liver Disease (NAFLD) is said to be 20-30% of the general population, and around 100% of obese groups [78]. The factors governing the progression to frank hepatic inflammation, known as Non-Alcoholic Steatohepatitis (NASH) were until recently unclear. The NLRP3 inflammasome is generally pro-inflammatory, whilst the NLRP6 (decoy) inflammasome is anti-inflammatory. Dysregulation of inflammasome activation was recently linked to an increased portal influx of TLR4 and TLR9 ligands into the hepatic circulation, and the triggering of liver inflammation and secondary metabolic dysbiosis [78]. The rate of flux of the gut derived ligands was linked to the rate of progression of NASH. NASH was felt to develop via a two step pathway with the first step being fat accumulation particularly in mitochondria, and the second frequently involving inflammation related oxidative damage [78]. Moreover two inflammasomes, NLRP3 and NLRP6 act as sensors and monitors of the colonic microflora. Disruption of these activities resulted in an alteration of the microflora with the adoption of colitigenic bacteria and downstream excessive inflammation [78]. The colitigenic bacteria were shown to be transferable to co-housed normal mice. Importantly the progression to NASH could be blocked either with the use of combined antibiotic regimes with ciprofloxacin and metronidazole, or the use of animals in which the TLR pathway was genetically blocked by abrogation of its downstream signalling components TLR5, MyD88, TRIF (TIR containing adaptor inducing interferon-β) or Asc (Apoptosis-associated speck-like protein containing a caspase recruitment domain) [78]. Levels of TLR4 and TLR9 ligands were found to be markedly increased in the portal circulation of the NASH mice. TNFα was shown to be a critically important mediator of the NAFLD-NASH transition, and gut derived CCL5 was shown to be a key mediator involved in its progression [78]. Indeed the rate of microbial induced colonic inflammation was shown to be the determining factor in the rate of progression to NASH. Stigmata of the metabolic syndrome including weight gain, elevated fasting glucose and insulin levels, and reduced glucose tolerance were all observed in the NASH animals, which was exacerbated by HFD feeding [78]. | |
| These data show that it is now established that the microorganisms of the bowel, far from being inert bystanders, are increasingly being viewed as actively interacting with the host both in normal development and maturation of the host immune system, with the induction of immune – metabolic disease, and even with epigenetic modification some of the major genes of host immunity. | |
Section VII: Appetite and Craving |
|
| A large and established body of research shows that the hypothalamus directs feeding and appetitive functions for the organism.This activity is centred mainly in the arcuate and paraventricular nuclei [317-326]. It is the counterbalance between the orexigenic AGRP/NPY and anorexigenic POMC/CART neurones which is known to control body weight and energy balance and appetitive drives for food, protein, glucose and fats. Indeed one recent elegant study was able to closely regulate feeding behaviour including inducing obesity by optical stimulation of neurons in the lateral hypothalamus which are in a state of tonic inhibition with those in the more medial locations [327]. | |
| POMC Neuronal Physiology and wiring | |
| The POMC neurons of the MBH have a number of interesting features. They have widespread outputs throughout the brain, although not the striatum, cerebral cortex, hippocampus or cerebellum [328]. Their neuroendocrine outputs fall into two classes, β-endorphin and the melanocortins ACTH, and α-, β- and γ- MSH, which are all breakdown products of POMC [4,37,329]. The peptide breakdown products from POMC catabolism are shown schematically in Figure 10. β-endorphin and ACTH are released into the peripheral circulation from the anterior pituitary gland. POMC neurons synapse onto GABA inhibitory interneurons, and receive inputs from these neurons also [328]. They output to dopaminergic neurons which in turn control GnRH releasing neurons. POMC neurons also receive dopaminergic inputs [38]. They also directly inhibit GnRH neurons in the lateral hypothalamus, the MBH and the preoptic area themselves [328,330]. POMC neurons are presynaptic targets of estrogens and androgens, which also modulate the inwardly rectifying K+-channels on POMC cells [329,331]. They also inhibit oxytocin and vasopressin neurons in the paraventricular and supraoptic areas [328]. POMC neurons also have CB1 endocannabinoid receptors which inhibit their activity [44,45]. | |
| Figure 10: Schematic of proteolytic breakdown products of proopiomelanocortin From Wikipedia, http://en.wikipedia.org/wiki/Proopiomelanocortin, viewed 1st September 2014. Public Domain. | |
| The melanocortins bind to melanocortin receptors (MCR) 1-5. MC1R is found in the skin and is concerned with expression of skin pigments. MC2R is the ACTH receptor in the adrenal gland. MC5R is expressed in various peripheral exocrine glands and regulates secretion. MC3R and MC4R are the central receptors of the appetite centre and mediate the signalling of satiety at numerous brain centres. MC3R is an inhibitory autoreceptor for the satiety circuit. Its action is thus very like that of the μ-opiate receptors (MOR) on POMC cells [38]. MC4R is found in over 100 brain centres, and is involved in diverse activities including food intake and energy expenditure, heart rate, blood pressure, inflammation, erectile function, female lordosis posturing and natriuresis. MC3R and MC4R are naturally antagonized by AGRP and neuropeptide Y (NPY), with many of the target neurons receiving inputs from both POMC and AGRP/NPY neurons (Figure 11). In addition to releasing inhibitory NPY and AGRP, AGRP/NPY neurons also release inhibitory GABA onto POMC neurons. Both GABA and NPY are released from the same AGRP boutons.10-15% of arcuate nucleus POMC neurons also have Y2 receptors for the orexigenic neuropeptide Peptide YY3-36 (PYY), which is related to NPY [329]. | |
| Figure 11: Mutual antagonism in arcuate nucleus between neuropeptide y and proopiomelanocortin neurons, from Crowley 2001 (Nature), used by permission. | |
| POMC neurons inhibit CRH (Corticotrophin Releasing Hormone) neurons and thereby play a key role in stress regulation [328]. LPS administration has been shown to stimulate anxiety like behaviours in mice [332]. The melanocortin α-MSH has been shown to have potent anxiolytic effects [67]. Importantly POMC neurons have on their surface β-endorphin and α-MSH MOR autoreceptors which auto-inhibit their own release, forming a negative feedback loop at the cell surface. This is an important system which is stimulated by the administration of exogenous opiates, which thereby inhibit and down regulate these cells (Figure 12) [38,328,333]. POMC resting membrane potential is -40 to -45 mV. Administration of an opioid results in a 10- 20mV hyperpolarization within 35-40 seconds, with a complete loss of spontaneous action potential firing [38]. Conversely opiate antagonists depolarize and therefore stimulate these cells [38-39]. | |
| Figure 12: Rapid depolarization of proopiomelanocortin neurons by opioid (met-enkephalin) from Crowley 2001 (nature), used by permission. | |
| The μ-opiate receptor is coupled via Gαi/o proteins to the activation of inwardly rectifying hyperpolarizing K+-channels, the inhibition of Ca2+-channels, or the inhibition of adenylcyclase [328]. | |
| The projections of these cells importantly include key areas well recognized to be involved in the mesolimbic reward system including the nucleus accumbens, the medial amygdala, the periaqueductal grey, and the ventral tegmental area [329]. Both the arcuate nucleus itself and the median eminence to which it projects are functionally outside of the blood brain barrier and are considered to be part of the circumventricular organs which are thereby exposed to the nutritional, hormonal and immune signals in the circulating blood [329,331]. | |
| Both POMC/CART and AGRP/NPY neurons have leptin and insulin receptors. Leptin is the main adipostatic hormone, and insulin subserves a similar function for the glucostat. Insulin also has some adipostatic action [329]. Leptin signalling under normal conditions is associated with signalling satiety. Leptin signals are transduced by STAT3 (signal transducer and activator of transcription 3) [334]. Leptin depolarizes POMC cells and increases their rate of firing [331], and rapidly over just a few hours, changes the number of synaptic inputs to POMC and AGRP cells such that the former are increased and the latter reduced [335]. Leptin rapidly causes the release of α-MSH from POMC cells to signal satiety [38]. Leptin decreases GABA release from inhibitory NPY neurons synapsing on POMC cells [331]. POMC neurons are also sensitive to IL-1β [331]. Leptin signals are normally opposed by SOCS3 (Suppressor Of Cytokine Signalling 3), a major transcription factor involved with controlling cytokine signalling [331,334]. Cytokine activity medicated by SOCS3 is a major source of leptin resistance [334]. Deficiency of POMC, MC3R, MC4R or leptin or its receptor are all associated with an obese phenotype [329]. | |
| One important study administered ciliaryneurotrophic factor (CNTF) to mice and was able to show substantial weight loss of about 15% which was sustained after the administration of CNTF was suspended [334]. This effect was in turn shown to be due to the ability of CNTF to induce neurogenesis in the MBH, particularly relating to its stimulation of the POMC cell number and activity. Importantly leptin signalling in the adult MBH POMC neurons of animals fed a HFD was deficient, but leptin signalling was normal in the newborn POMC neurons [334]. Hence a clear functional separation was shown between adult and newborn neurons. This work therefore has clear implications for conditions wherein hypothalamic neurogenesis is impaired. This important work has obvious parallels to opiate dependency where neurogenic defects, obesity, metabolic syndrome and increased fat mass are all well characterized (see Section II). This work was supported by similar work showing that adult hypothalamic neurogenesis was inhibited by high fat diet related to apoptosis of newborn neurons [336]. | |
| Like saturated fatty acids and IKKβ/NF-κB, opiates have been shown to inhibit neurogenesis in the medial temporal lobe [337-343]. | |
| Interestingly the MBH POMC neurons have output to the nucleus of the solitary tract (NTS) which is the primary site of origin of the efferents for the vagus nerve to the gut. The NTS has a viscerotopic map of the GIT from its rostral to its caudal aspect [329]. This brain stem nucleus contains about 10% of the POMC neurons found in the MBH [38]. | |
| The above considerations show that perturbations of POMC physiology and regulation as are induced either by opioidergic neuronal hyperpolarization of immune-mediated POMC neuronal stress, injury, damage or apoptosis, must of necessity modulate the host of secondary and downstream pathways which depend upon it. | |
| Section VII: Stem Cells and Their Promoters | |
| One important focus of drug related toxicological studies has been stem cells. Classical studies dating from to 1970’s showed that opiate administration was associated with inhibition of tissue growth [344-363]. That this growth retardation could not only be prevented but be quantitatively reversed by the administration of opiate antagonists (racemic) naloxone and naltrexone suggested that indeed tissue growth was under tonic opioid inhibitory control [364-375]. The extent of the growth inhibition and stimulation was about 20- 30% in many of these studies. All organs studied were affected. Many studies were devoted to the structural and functional effects on brain growth with several studies looking at brain subregions. Much of this activity was subsequently shown to be related to met-enkephalin activity, which the authors named Opioid Growth Factor (OGF). However rather than acting at classical plasma membrane opiate receptors, OGF was shown to act at a cytoplasmic receptor located in the perinuclear space, which was duly cloned and sequenced, and its gene identified and sequenced, and denoted as OGFr [26]. The activities of OGF-OGFr were shown to be conserved across all tissues examined, many species from microbes to men, and all tissues in health and disease and including many human malignancies [26]. OGF-OGFr acts by imposing a block on the cell cycle progression through P16 and P21 in both normal and many malignant cell lines [376]. These findings imply that there is a direct pathway by which exogenously administered opiates can directly affect tissue growth which is particularly relevant at key stages of development including the in utero and adolescent growth spurts. This makes sense of the uniform findings of opiate induced tissue growth inhibition in essentially all tissues. | |
| Tissue stem cells are believed to function to maintain the health of organs and ultimately the organism. Most tissues have stem cell niches which nurse the stem cells and provide a satisfactory microenvironment for them in which to operate. The niches are believed to provide not only sustenance to the stem cells and their progeny, but also important local and systemic signals which govern their behaviour [32,377-383]. Stem cell numbers and activity are not static, but can be induced by various signals emanating from their local environment or brought to them in the circulation [384]. Stem cells are morphologically different in different tissues and have different genes activated consistent with that tissue and its function, and the developmental stage at which they occur. These disparities imply that it is not possible to consider tissue specific stem cells as one homogenous group. | |
| However many of the tissue specific stem cells express the embryonic stem cell genes Oct4, Nanog, Sox 2 or Myc. Indeed the key transcription factors which have been shown to be associated with somatic cell re-programming back to pluripotency are Oct4, Sox2, Klf4 and Myc (OSKM) [317]. | |
| With the exception of haemopoietic stem cells [32,381,385] and the stem cells of the pituitary gland [386,387] both of which are involved in the acute response to stressful stimuli, most stem cells are powerfully suppressed by immune activity [388-401]. Both Oct4 [402] and Nanog [403] have cytokine response elements in their promoters. Most such promoters are designed to receive signals transduced by STAT3 which is one of the canonical transcription factors involved in regulating stem cell activity [404,405]. The gp130 cytokines (glycoprotein 130) family including IL-6 and Leukaemia Inhibitory Factor (LIF) also signal to STAT3 and bind to similar cassettes, and form one of the major ways by which stem activity is regulated [402]. Moreover cross-activation of transcription factors also occurs, as Nanog activation is also dependent on binding of Oct4 and Sox2 [406]. Indeed Oct4 can heterodimerize with Sox 2 and can form feed forward and autoregulatory loops. | |
| Nanog also has a Tcf (Transcription Factor 7) / Lef (Lymphoid Enhancer Factor) response element and a SMAD (Small Body Size - Mothers AgainstDecapentaplegic) response element to receive input from TGFβ signalling. | |
| The Tcf/Lef response element implies that Nanog can be sensitive to β-catenin signalling which is a very important signal in both development and oncogenesis. Indeed the proximal Tcf/Lef site was noted to be critical to Nanog expression [407]. Indeed Oct4, Nanog and Sox2 have been shown to bind to promoters of members of the TGFβ and Wnt - β-catenin signalling pathways [406]. | |
| Oct 4 achieves epigenetic regulation in several ways. One of its target genes is FOXO1A, a member of the forkhead family of transcription factors which is involved in metabolic regulation [406]. Oct4, Sox2 and Nanog were associated with the promoters of 14 miRNA genes, and co-occupied promoters for mir-137 and mir-301. They also direct the activity of major morphogen programs such as the polycomb-trithorax groups [384]. | |
| Interestingly TLR4 has been shown to be an important negative regulator of adult neurogenesis in the hippocampus [244]. Contrariwise, the opiate antagonist naltrexone has been shown to block endotoxic shock by inhibiting TNFα [281]. | |
| The major point to be considered from this brief overview is that opiates themselves directly impede tissue growth both in the adult and at key developmental stages through well described pathways impacting stem cell growth. Moreover stem cells carry response elements in their promoter which respond specifically to gp130 cytokines in the IL-6 family, and these form one of the major regulating factors on stem cell activities. In most tissues the impact of cytokine stimulation is strongly and rapidly inhibitory. Given that opiates have been shown to be immunostimulatory, this implies that stem cells are subjected to a triple insult: directly from OGFOGFr mediated cell cycle arrest; through immunological insult; and by the interactive effects of the particular susceptibility of stem and progenitor cells to cytokine induced inhibition. | |
Section IX: Newer Treatments |
|
| There are two new forms of treatment which are relevant to this discussion.The first relates to a new form of delivery of the opiate antagonist naltrexone which has the ability to circumvent many of the concerns relating to compliance and so safety and overdose which had been felt in relation to the oral formulation of the agent. The second form of new and exciting treatments relate to the down regulation of the central neuroinflammation described in Section IV.In particular since TLR4 is widely believed to be a key regulator of this system, and intensive effort is going into modulation of the TLR4 signalling pathway, its binding partners, its scaffolding proteins, and the downstream signalling transduction pathway.IKKβ/NF-κB is similarly an important therapeutic target. | |
| Naltrexone implants | |
| Formerly voiced programmatic concerns relating to oral forms of naltrexone typically related to the acceptability of antagonist treatments to drug dependent patients, compliance issues with oral formulations of the agents, and overall safety and efficacy concerns [408-411]. 7 clinical trials of naltrexone implants [412-419] and a number of recent reviews [34,418] suggest that these challenges have been successfully addressed. In considering these reviews it should be noted that the authors from Norway in the last mentioned paper were also the authors of the last Cochrane review of the subject, and are also presently understood to be working on the present revision of that review, which has been stimulated by the flurry of recent very positive clinical trials in this area. | |
| Indeed registration of such slow release formulations in both the USA and in Russia, suggest that we are on the very threshold of a new treatment paradigm. It implies that reliable, safe, acceptable antagonist treatments are about to become a reality for significant numbers of patients dependent upon a range of chemical substances [39,414,420-436]. Naltrexone (and naloxone) have been noted consistently throughout many studies not only to reverse both the stem cell defects and the immune dysfunction (stimulation / suppression), but to positively modulate these changes. This is reflected in the very positive outcomes of mortality studies of the use of these implantable agents [437-439]. | |
| Suppressors of central inflammation | |
| TLR4 activation has been shown to be critically involved in many diseases including necrotizing enterocolitis, abdominal sepsis, arthritis, pneumonia, atherosclerosis, pancreatitis [284], chronic periodontitis [301,440,441] osteoporosis [442-445], emphysema [446], dementia [447-451] (including TLR7), alcoholic liver injury [76,452], hypertension [270] and drug dependence [25,242,255,257]. Presently existing drugs which target this system include ibudilast, minocycline, propentofylline, and pentoxifylline [25,93,242,257], but their shortcomings include poor efficacy, the inability to administer adequate doses therapeutically, or a challenging side effect profile. | |
| Ibudilast | |
| Of these agents, known as glial modulators, ibudilast (AV411) is the agent presently under most intensive investigation. Indeed review of the clinicaltrials.gov website shows that whilst no trials are registered of the latter three agents there are 7 clinical trials presently logged on the site for ibudilast. Four of these are presently recruiting patients.There are two trials for opiate dependency (oxycodone abuse and headache medication overuse), one trial in opiate withdrawal, one in chronic migraine, two in amphetamine abuse (one of which is also studying HIV infection) and one in multiple sclerosis. | |
| Ibudilast is marketed in the USA by a company called Medicanova under licence from a parent company in Osaka Japan. Ibudilast is available in Japan, Korea, Thailand and Philippines. The company has a licence to explore higher dose capsules than the 10 and 15 mg capsules commonly used in asthma. Present trials are using 50mg b.i.d. slow release formulations. Medicanova releases note that ibudilast is a glial modulator which suppresses proinflammatory cytokines such as TNFα, IL-1β and IL-6 and up-regulates anti-inflammatory IL-10 and neurotrophic factors. Hence it has anti-inflammatory and neurotrophic actions, and is therefore being explored in chronic pain syndromes and progressive multiple sclerosis [453]. | |
| Comments from Prof Sandra Comer of the New York Psychiatric Institute at Columbia University on the clincialtrials.gov website (http://clinicaltrials.gov/show/NCT01740414 viewed 6th November 2013) note that recent Columbia University studies demonstrate that ibudilast reduces the symptomatic discomfort of patients withdrawing from opiates. The abstract from this group presented at the fall San Diego American academy of Neurology meeting noted that the trial was small with three groups each of 10 patients in a placebo, and a low and high dose ibudilast group [454].The study was conducted over three weeks. No significant side effects were seen, and no discontinuations due to treatment. Dose related effects of ibudilast were seen with the highest dose (40 mg b.i.d.) reducing the subjective opiate withdrawal scores, and provided significantly improved opiate analgesia in opiate dependent patients. Moreover opiate induced pupillary constriction (miosis) was more marked in the patients in the higher dose ibudilast treatment group, consistent with lesser tolerance to the administered opiates [454]. | |
| One notes that the next study planned by this group will use a dose of 50mg bid., presumably indicating that the investigators feel that the drug is safe, and since a dose related response was found, the researchers are keen to study higher doses [453,455]. | |
| Similarly a recent article in the Huffington Post 4/03/2013 reported a preliminary UCLA study into 11 non-treatment seeking methamphetamine addicts [456]. The drug was safe, eased the addiction, made patients more rational, reduced craving, and improved cognitive function, often significantly.Patients given the drug seemed to find it much easier to resist the urge to use the agent. A second phase of the study is expected to commence in the middle of the US summer, and recruit 140 patients. It was mentioned to be the only pharmacological treatment for amphetamine addiction [456]. | |
| Hence there is clearly great excitement in the US about this agent. Indeed Medicanova is a small company with only 15 employees, and 85% of their research in these seven trials is being funded by NIDA (National Institutes of Drug Abuse, one of the National Institutes of Health (NIH) of the US Government), and the compound has been given expedited status by the US Food and Drug Administration (FDA) because of the potential of the agent and the enormous need for its various important applications [453]. | |
| TLR4 antagonists | |
| Several structural analogues of the lipid A segment of endotoxin have been synthesized which bound tightly to the large internal binding pocket of the TLR4 binding partner MD-2 and block TLR4 signalling in enterocytes and macrophages, and were able to block the development of experimental systemic endotoxaemia and necrotizing enterocolitis [284]. These are designed to function as TLR4 inhibitors. The lead compounds were known as C34 and C35.Inhibitors of other key steps in the TLR signalling pathway are also being developed, particularly MyD88, which would have similar function to TLR4 antagonists themselves. | |
| The Adelaide-Colorado-Maryland group is also understood to be intensively investigating the field of TLR4 antagonists. | |
| PTI-609 is the lead compound developed by Pain Therapeutics Inc. The story of its development is particularly fascinating [457]. Ultralow dose naltrexone administered in the femto- nano- molar range had long been known to potentiate morphine analgesia, although its molecular basis was unclear. These workers noted earlier work which suggested that part of the therapeutic switch from opiate analgesia to opiate induced hyperalgesia involved a switch in the intracellular coupling of the MOR from Gαi/o to Gs coupling to adenylcyclase, using Gβγ components derived from Gs proteins. It was this switch which was felt to make the MOR stimulation stimulatory rather than inhibitory. Since naltrexone interacts with the MOR at concentrations in the femtomolar range well below its binding affinity for the MOR proper, they searched for nearby binding sites which might be expected to affect the MOR by examining the protein-protein interactome of the MOR. They identified such a high affinity binding site on filamin A with an IC50 at 3.94 picomolar. Filamin A is a structural protein which interacts with about 30 different cell surface receptors including the MOR. Their lead compound PTI-609 has been shown to be strongly anti-inflammatory, and non-addictive. It carries the unique advantage of being both a potent analgesic, and potently anti-inflammatory combining the advantages of both systems. As such it is an important proof-of-principle agent [457]. | |
Section X: Clinical Implications |
|
| The above data have several major clinical implications. Whilst the following list is not exhaustive, it is intended for illustrative purposes. | |
| 1) Anxiety / Depression | |
| 2) Withdrawal / Tolerance / Dependence | |
| 3) Constipation | |
| 4) Congenital organ and brain damage | |
| 5) Cancer induction | |
| 6) Long term treatments – Morbidity Profile | |
| 7) Long term treatments – Mortality Profile / Longevity Considerations | |
| 8) Central anti-inflammatory agents | |
| 9) Antagonist treatments preferable to agonists | |
| Anxiety and depression | |
| Both these disorders in their more severe forms are seen as having an important neuroinflammatory organic substrate. Biomarkers of this central inflammation have been defined in the peripheral serum in many studies [458-473], and also centrally [474-478]. Similarly exorbitant rates of anxiety and depression have been noted in many clinical series of drug dependent patients such that such conditions can be said to be almost holoendemic in some long term clinics [11,477,479,480]. Given the now well established neuroinflammatory actions of most drugs of abuse it is no longer possible to argue that such as association is simply random. Whilst the argument that patients with pre-existing mood or other psychological disorder are more likely to use drugs has in part been substantiated, it is no longer scientifically sound to claim the long term self-administration or therapeutic administration of addictive agents is neuropathologically benign. | |
| Withdrawal / Tolerance / Dependence | |
| The neuroinflammatory contribution to on-going drug dependence has been shown many times in several elegant studies, particularly from the Adelaide-Colorado-Maryland group, as noted in Section IV. This has a very interesting implication for the fundamental understanding of patients who find themselves dependent upon depressant agents such as alcohol and opiates. This means that during their withdrawal phase, when the sedative effects of their agents are wearing off, the underlying neuroinflammatory actions of these agents will be acting unopposed and will become unmasked. This unmasked neuroinflammation will manifest clinically as agitated and anxious behaviour. In the drug dependent patient’s frame of reference the major effective sedative to which he is most accustomed is of course his primary drug of abuse. This provides a potent stimulus to reexposure to the drug to self-treat the aversive withdrawal syndrome, frequently characterized by hypercytokinaemia (short of “cytokine storm” [385,481-486] and heightened sympathoadrenal activation. This then provides the trigger to re-abuse in the short term daily cycles, irrespective of what may appear to an external observer to be clear evidences of severe consequences of such abuse. | |
| However it is also critically important to understand that such a dynamic will be operating on every time scale. Whilst this dynamic is true of the daily cycles of abuse and withdrawal, it is also true of regimes which attempt to slowly taper the therapeutic drug of abuse, such as may be undertaken with gradual methadone or buprenorphine reduction programs. That is to say that one of the major reasons for the failure of gradual agonist reduction programs is the emergence of severe and disabling anxiety as a particular lower threshold dose is reached. Hence every time a treatment dose is lowered to such a threshold point (which will be different for each patient), the neuroinflammatory excitation becomes clinically significant and problematic anxiety disrupts the previously planned reductions. Whilst alternative sedatives may be chosen the clinical reality is that patients will often insist upon the sure “cure” associated with reversal of the previous dose reduction. | |
| Furthermore since it may be that the central neuroinflammation is worse after protracted drug exposure than prior, it may be that once the agonist is withdrawn completely, an underlying significant psychological illness becomes unmasked which requires to be treated variously with anxiolytics, mood stabilizers, or antipsychotic agents. It is therefore paradoxical that what frequently begins with experimentation with mood altering drugs, becomes a fixed and problematic psychopathology as the induction or exacerbation of central neuroinflammation gives rise to a fixed neuropsychological disorder which required on-going treatment and therapy. | |
| Furthermore since inflammatory processes contain many positive feedback amplificatory loops within them, it is feasible that the inflammatory circuits which have been established during drug dependence do not necessarily cease once the drug of exposure is withdrawn. Indeed studies exist which clearly document that cessation of drug use is associated with a long term continued exacerbation of the underlying neuroinflammation over at least one or several years [110]. Since neuroinflammation is associated with drug craving, this explains the observation that many patients describe being “tortured” by persistent thoughts of using against their conscious will in many respects, and often including vivid and very disturbing drug using dreams and severe panic attack like thoughts of using and relapse. Indeed given the feed forward self-amplificatory loop nature of many inflammatory processes which may naturally progress without further external stimulation, it may be that the neuroinflammatory component of the dependence actually escalates during the drug free period post-detoxification which has the effect of making relapse more common with elapsed time rather than less so. This explains the common pattern seen in addiction where relapse becomes more common with time elapsed in sobriety, rather than less so as might otherwise be expected. | |
| It also explains why patients treated with stable forms of opiate antagonist treatments, such as naltrexone implants, describe an at first rapid, and then protracted freedom from such perseverative thoughts, no doubt related to the well described effect of classical opiate antagonists to suppress central neuroinflammation, which incidentally is not seen with naltrexone congeners which are impermeable into the CNS [255,489]. Indeed this effect is experienced very rapidly by many naltrexone implant patients within days, and over 90% have been reported to experience the extinguishment of craving after six weeks of oral treatment [488,489]. | |
| Constipation | |
| Constipation has always been a well described feature of opiate dependence since classical times [79]. Importantly delayed intestinal transit is a feature of ageing [293]. Moreover it is one feature to which patients often do not develop significant tolerance [79-80]. It may become associated with disorders of colonic motility.The above considerations suggest that the implications of the chronically loaded colon are likely to be associated with an increased flux of TLR ligands and endotoxins to the liver through an often leaky mucosal and blood-lumen barrier [76-78]. The common incidence of liver disease and partial portal shunting in these patients implies that this increased flux of TLR ligands into the liver may be associated both with heightened Kupffer, stellate and endothelial cell activation in the liver itself, and the release of an increase flux of TLR ligands and cytokines and chemokines into the systemic circulation. From the blood these antigens, cytokines and immune active cells can easily access the medial hypothalamus through a variety of pathways, not least of which include the circumventricular location of the median eminence and the arcuate nucleus described above. That is to say that the well described and very refractory constipation often seen in these patients is not benign, particularly over the longer term. | |
| Congenital organ / Brain damage | |
| Since opiates are immune active agents, and since these immune proteins are known to be very involved in morphogenesis and body patterning processes in a conserved manner from the fly to mammals, an elevated rate of congenital abnormalities in drug exposed infants is not entirely unexpected. This is particularly true of brain malformations whose pathogenesis was described above, and particularly implicates activities by MHC class I, C1q and C3 complement fragments, TNFα, neuronal pentraxins and the TLR’s. | |
| Cancer | |
| The association of inflammation with tumourigenesis is a now extensively researched subject. Indeed a recent PubMed search revealed 45,375 hits of this topic; 1,332 papers on the link between TLR’s and cancer; and 285 review papers on the TLR-cancer association alone (viewed 2nd November 2013).Many papers describe this link in relation to the following tumours: leukaemia, lymphoma, breast, prostate, gastrointestinal and other malignancies [490-506]. The strongly pro-oxidant inflammatory environment has been noted above [507]. Such free radicals are known to damage DNA in a mutagenic manner, and also critical thiol groups in many proteins including key tumour suppressor proteins such as P53 [508-510]. The now well described association of cancer therefore with drugs of addiction as detailed in Section II cannot be continually ascribed to solely to extraneous factors as has often been suggested [144,511]. Indeed numerous pathophysiological pathways now account for such changes, and that is without considering epigenetic impacts or telomere dysfunction as have been mentioned at various points above. | |
| Similar considerations apply to congenital cancers occurring in infants exposed either in utero to maternal drug use, or potentially to the impact of paternal drug use on the male gametes prior to conception. | |
| Long term morbidity profile | |
| The effect of immunostimulation and its contingent immunosuppression, stem cell suppression and the interaction of the two can reasonably be expected to take a significant toll on organ health across the organism particularly over the longer term in a kind of compounding interest type scenario. It suggests that both single organ and multiple organ dysfunction will be more common, which as noted in Section II, is the disease profile which has now been observed and quantitated. | |
| This is exacerbated by several factors. The first is the appetitive perturbation produced in the hypothalamus by drugs of dependence. As noted this is usually for a calorie dense diet high in saturated fats and simple carbohydrates.This tendency is exacerbated by diversion of resources which typically occurs away from nutritious foods to fast food and general undernourishment and malnourishment. Thirdly the metabolic syndrome which is induced by both peripheral and central inflammation tends to exacerbate the effects of dysglycaemia, insulinand leptin- resistance dyslipidaemia, over-nutrition and increased fat mass which may accompany many chemical dependencies such as methadone [129,130,137]. Moreover direct interaction between fatty acids and the NLRP3 inflammasome has been demonstrated with unsaturated fatty acids shown to prime the NLRP3 inflammasome via TLR4 ligation in dendritic cells with associated induction of insulin resistance [274]. | |
| The corollary of this is that the long term administration of drugs of abuse, even if undertaken in a therapeutic context, cannot be considered to be benign. This is in contradistinction to common messages which have been sent in earlier times [80,512-517]. The clear clinical message is that both the dose and duration of any such treatment should be minimized to avoid the accrual of long term end organ disease, albeit that much of that disease will remain subclinical for protracted periods of time as major clinical disorders often have very long incubation periods, even in drug dependent patients. | |
| Long term mortality profile / Longevity considerations | |
| Addictive drugs have been shown to induce neuroinflammation in the hypothalamus which not only coordinates much homeostatic, metabolic and hormonal regulation but has now been shown to coordinate lifespan [59]. This implies that the process of drug addiction directly impinges upon lifespan determination, not only by inducing elevated rates of damage on peripheral and central tissues by way of stem cell and inflammatory insults and their interactions, but also directly on one of the body’s main ageing directing centres. This suggests that progeroid processes not only underlie the observed elevation organ system mortality, but that centrally directed ageing – accelerant forces are also at play to speed the development of premature and multisystem disease. This suggests that the elevated rates of death seen in these patients is related not just to the accrual of organ specific disease, but to an acceleration of the ageing process itself, as has now been observed and quantitated [19,20,22,110,518]. | |
| Again the clear demonstration that measures of biological age have been repeatedly related with high significance to the dose and duration of total opiate exposure [8,13,19,20,22,518] confirm the importance of trying to limit as far as possible the extent to which patients are exposed to opiates either as part of their selfadministration and lifestyle, or as part of their therapeutic regime. Elevation of biological age implies acceleration of ageing processes and premature intrinsic (non-overdose related) mortality. Indeed in some studies the biological age was best predicted by the square of the duration of opiate administration [518] further underscoring the importance of minimizing the period of opiate exposure to optimize long term health and presumably mortality outcomes. In this context the exacerbation of the acceleration of biological ageing with high dose full opiate agonists [20], and the reversal of this process with the assistance of nonspecific opiate antagonists [36], is of particularly interest. | |
| Central Anti-inflammatory agents | |
| The work on central TLR4 antagonists and other means to block the pathway from lipoprotein binding protein (LBP) – CD14- MD2 – TLR4 to IKKβ/NF-κB activation suggests that small molecular inhibitors of this pathways are likely to become available in the near future. Moreover, as in the case of PTI-609 it may be that such agents can be combined into analgesics active at the μ-opiate receptor. The advantages of such compounds to a host of neurological disorders are likely to be many fold, as have been clearly expounded by the work of the group of Hutchinson and Watkins [519,520]. This will likely include less habituation, tolerance and dependence, lower rates of overdose, improved metabolic profile, improved neurogenesis in many brain regions, reduced chronic pain syndromes, and improved psychiatric status. The very fact of the existence of an energetic drive for the discovery of new candidate molecules clearly indicates that there is a widespread appreciation of the importance of exploring such pathways. | |
| Treatment choice of agonism v. antagonism | |
| These considerations further imply that antagonist treatments may be preferable to agonist treatments provided certain programmatic shortcomings can be adequately addressed. As discussed in the preceding section, the imminent availability of implantable reliable and safe forms of depot naltrexone suggest that this option will become an increasingly important and likely more physiological choice for patients than the present balance of treatments which is weighted more in favour of agonist based treatments in many nations [415,417,521]. | |
Conclusion |
|
| It is clear that research conducted in disparate fields far beyond what is typically considered the realm of addiction medicine will have a radical and transformative influence on the future of addiction treatment. Exposure to exogenous opiates has been shown to be associated with metabolic syndrome, hyperglycaemia, insulin resistance, hypertension, weight gain, inflammation-related psychiatric disorders, many physical disorders, accelerated ageing and elevated mortality related to every major organ system; and opiateinduced hypothalamic inflammation has been shown to be causally related to the induction of disease and systemic ageing processes. This has implications for the duration for which opiate agonist therapies are recommended, and indicate more energetic efforts to bring alternative treatments into mainstream clinical practice. | |
References |
|
|
|