Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Pharm Sci Emerg Drugs Vol: 6 Issue: 1
High-Throughput Structure-Based Drug Design of Chalcones Scaffolds as Dual Inhibitor of Cyclooxygenase-2 and Microsomal Prostaglandin E Synthase-1
Muhd Hanis Md Idris, Siti Norhidayu Mohd Amin, Manikandan Selvaraj, Hisyam Jamari, Teh Lay Kek and Mohd Zaki Salleh*
Integrative Pharmacogenomics Institute (iPROMISE), Universiti Teknologi MARA (UiTM), Selangor, Malaysia
*Corresponding Author : Mohd Zaki Salleh
Integrative Pharmacogenomics Institute (iPROMISE), Universiti Teknologi MARA (UiTM) Selangor, Puncak Alam Campus, 42300 Bandar Puncak Alam, Selangor, Malaysia
Tel: +603-3258 4685
Fax: +603-3258 4658
E-mail: zakisalleh.ipromise@gmail.com
Received: June 05, 2018 Accepted: June 21, 2018 Published: June 28, 2018
Citation: Md Idris MH, Amin SNM, Selvaraj M, Jamari H, Kek TL (2018) High-Throughput Structure-Based Drug Design of Chalcones Scaffolds as Dual Inhibitor of Cyclooxygenase-2 and Microsomal Prostaglandin E Synthase-1. J Pharm Sci Emerg Drugs 6:1. doi: 10.4172/2380-9477.1000128
Abstract
Cyclooxygenase-2 (COX-2) inhibitors had been extensively used to treat inflammatory disorders. However, the increased incidence of cardiovascular side effects had limited the usage of the drugs. The emergence of microsomal prostaglandin E synthase-1 (mPGES-1) as newly recognized therapeutic target for inflammation and pain gave new hope to develop anti-inflammatory drug with minimal adverse effects. Thus, selective inhibition of COX-2 and mPGES-1 is expected to provide anti-inflammatory effects without the side effects. Chalcone is a compound derived from nature with various pharmacological activities. Based on its favorable activity, chalcone framework had been used to identify novel derivatives for anti-inflammatory properties. Hence, the present study aims to identify chalcone derivatives that can reduce the production of the inducible PGE2 by targeting COX-2 and mPGES-1 using structure-based drug design. A hybrid pharmacophore model of COX-2 named as “phore 1” was developed and used to screen chalcone derivatives from ASINEX database. Forty-two compounds were successfully mapped on “phore 1” and only 15 compounds were selected based on docking. These compounds were then subjected to pharmacophore screening and followed by docking for mPGES-1. Three lead compounds (BAS00384673, BAS00643043 and BAS02557914) were found to be selective towards COX-2 and have good binding interaction with mPGES-1.
Keywords: COX; mPGES-1; Pharmacophore; Flavonoids; Virtual screening
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used in treating pain and inflammation through the inhibitions of either COX-1 and COX-2 enzymes or specifically COX-2 alone [1,2]. However, inhibition of the production of PGE2 due to inhibition of COX-1 as in traditional NSAIDs (tNSAIDs) such as ibuprofen, indomethacin, diclofenac, ketoprofen, naproxen, piroxicam and nabumetone are associated with gastrointestinal bleeding [3,4]. In early 1990s, the discovery of COX-2 enzyme had led to the development of new generation of selective NSAIDs (COX-2 inhibitors). Even though COX-2 inhibitors (COXIB) had shown reduced incidences of gastrotoxicity these inhibitors had been associated with cardiovascular problems such as high blood pressure, atherogenesis and heart attack [5]. These side effects were due to the inhibition of other inducible prostanoids such as prostacyclin (PGI2) [3,6]. In fact, overexpression of COX-2 is associated with the pathogenesis and progression of inflammatory diseases such as cancer, rheumatoid arthritis, diabetes, sclerosis, atherosclerosis, and chronic obstructive pulmonary disease [7,8]. Due to the adverse effects of selective COXIB, new inflammatory targets were explored and studied including PGE2 synthase (PGES). PGES regulates the final step of the biosynthesis of PGE2 in arachidonic acid pathway. Interestingly, inhibiting PGES can reduce the production of PGE2 without affecting the generation of other PGs and thromboxane (Tx) and consequently result in fewer side effects. In general, PGES is divided into three types: cytosolic PGE2 synthase (cPGES) and two (2) microsomal PGE2 which are referred to as mPGES-1 and mPGES-2 [6]. Among these three (3) types of PGES, mPGES-1 is the major enzyme responsible for the biosynthesis of inducible PGE2 during inflammatory response and this enzyme is distinctly different structurally and biologically from other PGES. Recent research reported that common adverse effects of NSAIDs would be avoided by inhibition of mPGES-1 [9]. In fact, deletion of mPGES-1 in mice caused less thrombogenesis or low blood pressure in normolipidemic mice compared to the deletion of COX-2 gene [10]. Thus, selective dual-inhibition of COX-2 and mPGES-1 had potential to be developed as anti-inflammatory and analgesic agents with reduced adverse effects. Chalcone which belongs to the flavonoid family is commonly found in plants such as fruits, vegetables, spices and tea. This compound has been demonstrated to exhibit a variety of important biological activities including anti-cancer [11,12], antiviral [13], anti-fungal [14], anti-microbial [15], anti-malarial [16,17], anti-diabetic [18], anti-oxidant [19], anti-inflammatory [20,21] and antinociceptive [20]. In a recent study, chalcone and its derivatives were reported to suppress the proliferation of MCF-7 cells by modulating pro-inflammatory markers [22]. Chalcones were shown to have anti-inflammatory activities in acute lung injury and had provided protection against hepatic injury by inhibiting hepatic fibrosis and inflammation [23,24]. Furthermore, our preliminary study showed that chalcone derivatives exerted reduction of inflammatory-mediated pain in rats subjected to formalin test [25]. In continuation of our previous study, the present study was designed to identify compounds that reduce the production of inducible PGE2 by targeting COX-2 and mPGES-1 based on chalcone scaffold. This study used structurebased drug design approach which involved screening of the hybrid pharmacophore and followed by molecular docking. To the best of our knowledge, the approach using hybrid pharmacophore of COX-2 together with targeting dual-inhibitor of COX-2 and PGES-1 had not been reported yet. The approach was designed to increase the chances in finding good inhibitors with less undesirable side effects.
Materials and Methods
Generation and validation of pharmacophore for COX-2 inhibitors
Structure-based pharmacophore model of COX-2 inhibitors were generated using LigandScout software (Inte: Ligand GmbH, Austria). Six (6) COX-2 structures bounded with different active ligands were extracted from the Protein Data Bank (PDB) (PDB ID: 3LN1, 3LN0, 1CX2, 6COX, 4OTY, 3MQE) (http://www.rcsb.org) and were used to develop COX-2 structure-based pharmacophore model. The pharmacophoric features such as hydrogen-bond acceptor (HBA), hydrogen-bond donor (HBD), hydrophobic (HY), negative ionizable (NI), positive ionizable (PI) and aromatic ring (AR) were manually checked based on the reported information as described in literature and PDB sum (http://www.ebi.ac.uk/pdbsum).
For 1CX2 and 6COX, same inhibitor (SC-558) was bounded to both COX-2 enzymes but by different binding modes [26,27]. Therefore, common pharmacophore features were generated from these PDB complexes. 1CX2 and 6COX pharmacophore features were aligned together using LigandScout (version 4.1 Advanced, Inte: Ligand GmbH, Maria Enzersdorf, Austria) and their shared pharmacophore features were generated. These common pharmacophore features were further used to generate COX-2 structure-based pharmacophore hypotheses. Several hybrid pharmacophore hypotheses were generated based on the common and merged pharmacophore features of each selected PDB complexes. The final pharmacophore model for COX-2 was name as “phore 1”.
The pharmacophore models were validated using a validation descriptor known as the enrichment factor (EF) [28]. The process of validation was performed using a test set of active and inactive COX-2 compounds from DEKOIS (http://www.dekois.com) which comprised of 40 active and 1199 inactive compounds [29]. The EF was calculated based on the formula below:
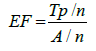
Where, TP is the number of active hits retrieved by pharmacophore (true positives), n is the number of active and inactive compounds found from pharmacophore hypothesis, A is the number of active compounds in the database and N is the database size (total of active and inactive).
Besides that, validation of pharmacophore was also performed based on pharmacophore fit value where the pharmacophore fit value for active compounds should be higher than inactive compounds (decoy set).
Generation of compounds library for virtual screening
Ligands that were used in this study were downloaded from ASINEX in SDF format which included Elite libraries, Gold and Platinum Collection (http://www.asinex.com). This dataset contains 523,376 lead and drug-like compounds. The compounds fulfilled Lipinski’s Rule of 5 by having molecular weight less than 500 Dalton with calculated LogP <5, number of hydrogen-bond donor’s ≤ 5-, and number of hydrogen-bond acceptors ≤ 10. The dataset of compounds was filtered based on chalcone scaffold using shape similarity search in Canvas (Maestro, Schrödinger, New York, USA). Then, the filtered ligands were converted to 3D structure and geometrically optimized, and energy minimized using LigPrep module (Schrödinger, New York, USA).
Virtual screening for COX inhibition
The initial virtual screening for COX inhibition was performed using the following step: (a) the compounds were screened using “phore 1” pharmacophore, (b) the compounds with fit values greater than 45.00 were considered for further testing, (c) molecular docking for COX enzymes -COX-1 and COX-2 were conducted, and (d) the compounds with docking score less than -8.0 kcal/mol for COX-2 were further evaluated in the second virtual screening.
Preparation of human COX-1 homology model: Human COX- 1 homology model was built as there was no crystal structure of human COX-1 available in PDB. The homology modeling of human COX-1 was performed using Prime tool (Schrödinger, New York, USA). The amino acid sequence for human COX-1 was retrieved from National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov). The sequence was used to search for the homologous proteins from the online PDB using protein-protein BLAST algorithm. The protein with the highest sequence identity was chosen as template for homology modelling of human COX-1. Next, the template and target sequence were aligned using Clustal W. The human COX-1 model structure with bounded ligand was developed using energy-based method. Later, the modelled structure was refined and minimized. The quality of modelled structure was subsequently evaluated using Ramachandran plot. The stability of the modelled structure of human COX-1 was determined using Desmond (Schrödinger, New York, USA). The parameters for molecular dynamics using Desmond were set at 15 ns based on previous study [30].
Molecular docking using GLIDE for COX
Human COX-1 homology model and human COX-2 structure (PDB ID: 5F19) were prepared using the Protein Preparation Wizard (Maestro, Schrödinger, New York, USA). The water molecules were removed, all hydrogens were added, and bond orders were assigned. The grid for both enzymes was generated by specifying the substrate in the active site (flurbiprofen for COX-1 and aspirin for COX-2) and the grid box was generated around the ligand with a grid spacing of 12 Å. Then, the hit compounds from “phore 1” screening were docked using GLIDE (Schrödinger, New York) in extra precision (XP) mode.
Generation and validation of pharmacophore model and validation for mPGES-1 inhibitors
A structure-based pharmacophore model for mPGES-1 inhibitors was developed based on the most potent mPGES-1inhibitor, 2-acylaminoimidazole (4UL) which co-crystallized in 5BQI using LigandScout (version 4.1 Advanced, Inte:Ligand GmbH, Maria Enzersdorf, Austria). The pharmacophore model was built based on the key intermolecular interaction of the compound (4UL). Similar validation method was used as described in Section 2.1 to validate the pharmacophore model of mPGES-1. Therefore, test set was developed based on 21 compounds (inactive compounds) that had been biologically tested but did not inhibit mPGES-1 and 10 mPGES-1 inhibitors (active compounds) were used in the training set. All active compounds of mPGES-1 inhibitors in the training set were selected based on the diversity of the structures for the chemical classes and were either acidic or non-acidic inhibitors of mPGES-1.
Virtual screening for mPGES-1 inhibition
The second virtual screening was performed to identify compounds which had binding interaction with mPGES-1. Firstly, the hit compounds with COX activity were virtually screened for mPGES-1 activity using structure-based pharmacophore model of mPGES-1 and it was followed by molecular docking. Hit compounds with dual-inhibition potentials were identified based on the criteria that the compounds have comparable results in pharmacophore fit as well as in docking score and have different binding mode in the presence and absence of GSH in mPGES-1 binding sites. The binding interactions of the hit compounds in the mPGES-1 binding site were visualized and analyzed using PyMOL (Schrödinger, New York, USA).
Molecular docking for mPGES-1
Monomeric X-ray complex structure of mPGES-1 with PDB ID: 4BPM was retrieved from PDB. Prior to docking, the protein structure was pre-processed using Protein Preparation Wizard (Maestro, Schrödinger, New York, USA). mPGES-1 existed naturally as homotrimer with three equivalent active site cavities for each monomer interface and it is located on the endoplasmic reticulum. Therefore, trimer structure of mPGES-1 was generated using the Crystal Mates feature in Maestro (Schrödinger, New York, USA). He and Lai et al. [31] suggested that an inhibitor of mPGES-1 could either occupy the binding sites of the PGH2 or partially displaces the cofactor of GSH. Therefore, in this study molecular docking for mPGES-1 protein was performed with GSH and without GSH. For docking without GSH, GSH was removed from binding site before docking was performed.
Binding sites of the mPGES-1 were located between the monomer. Therefore, mPGES-1 binding site was determined between chains A and B of mPGES-1. The grid size of the inner box was 16 × 26 × 22 meanwhile for the outer box was 28 × 38 × 34 and centered at -10.0557 (x), 16.623 (y), and 45.7128 (z), with grid spacing of 12 Å. Molecular docking was then performed using GLIDE in standard precision (SP) mode and extra precision (XP) mode according to Lauro et al. [32].
MM-GBSA calculation
MM-GBSA was calculated using Schrodinger software. The calculation was performed based on a single minimized proteinligand structure at target proteins of COX-1, COX-2, mPGES-1 with GSH and mPGES-1 without GSH.
Results and Discussion
COX-2 pharmacophore model generation and theoretical validation
Pharmacophore was built to outline the key intermolecular interactions needed for the drugs to inhibit COX-2. In this study, the features of inhibitors that contributed the most to the selectivity of COX-2 were identified. The pharmacophore generated were based on the diversity of chemical structures of COX-2 inhibitors which were SC-558, celecoxib, 5c-S, SC-75416 and lumiracoxib. As SC-558 was bounded in two different PDB complexes (1CX2 and 6COX), a common pharmacophore feature was generated to represent the pharmacophore feature of SC-553. Initially, pharmacophore features of SC-558 in both complexes consist of five HY and a HBD while two HBA and three HBA were generated in 1CX2 and 6COX, respectively. Two HBA features on trifluoromethyl group of SC- 558 in these pharmacophore models overlapped with HY feature. Therefore, the overlapping features of two HBA were removed to simplify the pharmacophore model. Then, the commonly shared pharmacophore features were derived from these PDB complexes as reported in Table 1. It comprised of five HY and a HBD features. The pharmacophore features of other inhibitors (celecoxib, lumiracoxib, 5c-S and SC-75416) were showed in Table 2. Several hybrid hypotheses were generated based on common and merged pharmacophore features of each inhibitor. The development of hybrid pharmacophore for COX-2 was completely new and we used a different approach in this study.
| PDB ID | Ligand | Pharmacophore of the ligand | Number of features | Pharmacophore features | Shared Pharmacophore features |
|---|---|---|---|---|---|
| 1CX2 | SC-558 | 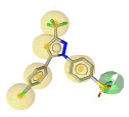 |
6 | 5 HY, 1 HBD | 5 HY, 1 HBD |
| 6COX | 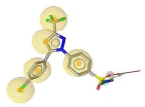 |
7 | 5 HY, 1 HBD, 1 HBA |
Table 1: Common pharmacophore features for SC-558.
| Ligand/ PDB ID | Chemical structure of ligand | Pharmacophore of the ligands | Number of features | Pharmacophore features |
|---|---|---|---|---|
| SC-558 |  |
 |
6 | 5 HY, 1 HBD |
| Celecoxib (3LN1) |  |
 |
6 | 5 HY, 1 HBD |
| 5c-S (3LN0) |
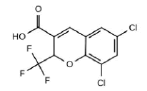 |
 |
7 | 5 HY, 2 HBA |
| SC-75416 (3MQE) |
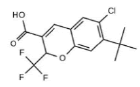 |
 |
7 | 5 HY, 2 HBA |
| Lumiracoxib (4OTY) |
 |
 |
8 | 6 HY, 2 HBA |
Table 2: Pharmacophore features for five selected ligands which were classified as COX-2 inhibitor.
Hypothesis 1 was generated by aligning all the chemical pharmacophore features of each inhibitors including the commonly shared pharmacophore features (SC-558). It comprised of four HY features which were required for the selectivity towards COX-2. Meanwhile for Hypothesis 2, two HBA features on the carboxylic group of the 5c-S, SC-75416 and lumiracoxib were considered. According to the binding interaction derived from the X-ray structures of these compounds in the COX-2 active site, the carboxylic group of each of the compounds (5c-S, SC-75416 and lumiracoxib) formed hydrogen bonds with phenol group of Tyr385 and hydroxyl group of Ser530 [33,34]. These two HBA features is important for selectivity of COX-2 even though other COX-2 inhibitors such as celecoxib and SC-558 (sulphonamide class) does not show any interaction with these amino acid residues (Tyr385 and Ser530). Therefore, the two HBA features were added to Hypothesis 1 which leads to generation of Hypothesis 2. Next, the shared pharmacophore features of 5c-S, SC-558 and SC- 75416 were merged with shared pharmacophore features between celecoxib and SC-558. As a result, hybrid pharmacophore model was generated (Hypothesis 3) which comprised of four HY and a HBA. A HBA feature of Hypothesis 3 was modified by increasing the tolerance radius at 0.15 Å. The generated pharmacophore hypothesis models were summarized in Table 3 and Figure 1.
| Pharmacophore hypothesis | n° of features | Chemical features of COX-2 pharmacophore model | Min n° of required features | EF | Rel EF | Actives found (%) (n=40) | Decoys found (%) (n=1199) |
|---|---|---|---|---|---|---|---|
| Hypothesis 1 | 4 | 4 HY | 4 | 1.51 | 0.049 | 32.5 | 21.1 |
| 3 | 2.52 | 0.081 | 85.0 | 31.8 | |||
| Hypothesis 2 | 6 | 4 HY, 2 HBA | 6 | 19.71 | 0.636 | 17.5 | 0.33 |
| 5 | 8.99 | 0.290 | 45.0 | 3.67 | |||
| 4 | 2.05 | 0.066 | 82.5 | 39.0 | |||
| 3 | 1.25 | 0.040 | 97.5 | 77.4 | |||
| Hypothesis 3 | 5 | 4 HY, 1 HBA | 5 | 0 | 0 | 0 | 0.08 |
| 4 | 2.14 | 0.069 | 5.0 | 2.25 | |||
| 3 | 2.21 | 0.071 | 15.0 | 6.51 |
Table 3: The structure-based pharmacophore hypotheses generation for COX-2.

Figure 1: Pharmacophore hypotheses for COX-2 that had been generated by LigandScout. (A) Hypothesis 1 for four HY features (yellow, a, b, c, d). (B) Hypothesis 2 for four HY (yellow, a, b, c, d) and two HBA (red, e, f). (C) Hypothesis 3 for four HY (yellow, a, b, c, d) and a HBA (red, f). Hydrophobic features (HY) were shown in yellow spheres and hydrogen bond acceptors (HBA) features were shown in red spheres or red arrows. Based on all the hypotheses, there are four common HY features (a, b, c, d).
All the hypotheses were validated based on the enrichment factors (EF) by implementing dataset which comprised of 40 active and 1199 inactive molecules of COX-2. The minimum number of selected features of each pharmacophore hypotheses of COX-2 were set at 3 and were validated based on the EF value (Table 3). Hypothesis 2 had the highest EF value which was 19.71 and the highest relative EF value of 0.636. It was chosen as the best pharmacophore hypothesis due to its relative EF, where the relative EF was calculated by dividing the EF with maximum EF. Hypothesis 2 consisted of six features (four HY and two HBA) (Figure 1); and demonstrated 17.5% probability of accuracy in predicting the active compounds in dataset and 1.4% of false results when Hypothesis 2 with the maximum number of required features were set.
Hypothesis 2 series were further evaluated to find the best pharmacophore hypothesis. In choosing the minimum number of required features in the pharmacophore hypothesis, the individual set of Hypothesis 2 was validated using the EF value. The active compounds identified using the different hypothesis were as follows: Hypothesis 2-6 identified 17.5% of active compounds (7 out of 40) whereas 40%, 82.5% and 97.5% were identified using Hypothesis 2-5 (18 out of 40), Hypothesis 2-4 (33 out of 40) and Hypothesis 2-3 (39 out of 40), respectively. Meantime, the percentage of false results for Hypothesis 2-6 was 0.3% (4 out of 1199), Hypothesis 2-5 was 3.67% (44 out of 1199), Hypothesis 2-4 was 38.9% (466 out of 1199) and Hypothesis 2-3 was 77.4% (928 out of 1199). Hypothesis 2 with the minimum number of required features of 4, was chosen as the final hypothesis parameter to screen the ASINEX database and was named as “phore 1”. Based on “phore 1” screening results of test and training set, the highest and lowest pharmacophore-fit score for COX-2 active compounds were 68.22 and 45.37 respectively. Therefore, the minimum cut-off of fit values in “phore 1” pharmacophore screening was set at 45.00.
COX structural analysis
Understanding the structures and the differences of COX isoforms is necessary in designing novel and selective COX inhibitors. COX-1 and COX-2 shared ~60% sequence identity and about 87% similarities for their active sites. COX active site comprised of long hydrophobic channel with narrow entrance at the membrane-binding domain [1]. Even though the active site is highly similar but its binding cavity is different where COX-2 has larger binding cavity than COX-1 which account for the differences in the selectivity of the COX inhibitors [1]. To identify the difference between COX-1 and COX-2, crystal structure with PDB ID of 1Q4G and 1CX2 (COX-1 and COX-2 respectively) were aligned together and compared. The active site of COX was divided into three regions: entrance of the active site which comprised of Arg120 and Tyr355; hydrophobic pocket which was beneath the heme group; and side pocket where the selectivity of the COX took place. The entrance of the active site and the hydrophobic pocket were highly conserved region, but the side pocket was nonconserved region where a few amino acid residues were different and gave rise to extra pocket to COX-2.
Extra side pocket in COX-2 is due to the difference in the single amino acid residue at position 523 which was near to Arg120 and it was important for selectivity of many drugs. The reduction of a single methyl group of isoleucine (Ile) in COX-1 to valine (Val) in COX-2 at position 523 leaves a space in the lining of binding site and allows drugs/inhibitors to access the side pocket [26]. The access of side pocket in COX-2 allows additional interactions with Arg513, substitution of His513 in COX-1, where the difference contributes to COX-2 selectivity. The orientation of Leu384 also contributes to the selectivity in COX-2. The presence of Phe503 causes the Leu384 side chain to point into the active site in COX-1 meanwhile in COX- 2, small size of Leu503 allows the Leu384 to move away from active site and to increase accessible space in COX-2 binding site. Besides that, substitution of amino acid residue at position 434 and 509 from isoleucine (Ile) in COX-1 to valine (Val) in COX-2 contributes to larger binding site in COX-2 compared to COX-1.
Based on the structural analysis of both COX enzymes, two factors played role in the selectivity of COX-2. Firstly, an inhibitor should occupy the side pocket in COX-2 by making Van der Waal forces and/or π-π interaction and/or polar/H-bond interactions with the residues that were lining within the side pocket. Secondly, the inhibitor should have polar/H-bond interactions with Tyr385 and/or Ser530. These interactions were important because the interactions were mainly observed in moderately COX-2 selective compounds such as diclofenac compared to other NSAIDs such as indomethacin, ibuprofen and naproxen. Further, Tyr385 and Ser530 residues were correlated in inhibiting COX-2 enzyme where Tyr385 stabilized the negative charge when acetylation of Ser530 occurs especially in NSAIDs such as diclofenac [35].
Preparation of COX structures
The crystal structure of human COX-1 is required to study and determine the selectivity of COX inhibitors. However, crystal structure of human COX-1 is not available in PDB database. Therefore, homology modeling was employed to build the structure based on the human COX-1 protein sequence available (Epstein et al. [36], Chothia and Lesk [37], Fiser [38]). In this study, human COX-1 homology model was constructed using complex structure of ovine COX-1 (PDB ID: 1Q4G) as the template with 93% sequence similarity with human and at resolution of 2.1 Å. At this level of sequence identity, only small residues were changed. The changes did not affect the secondary structure and active site of the enzyme as the residues located outside both area and thus this does not affect the binding of ligands in the docking study. Human COX-1 homology model was shown in Figure 2. The geometry of the final model of human COX-1 was evaluated using Ramachandran plot’s calculations computed in web-based bioinformatics tools, RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php). Human COX-1 Ramachandran plot was depicted in Figure 2. The result revealed that the model showed a good distribution of amino acid residues of human COX- 1 where 96.6% of the residues were in favorable region, 3.4% of the residues were in allowed region and 0% of the residues were in outlier region. The modeled structure of human COX-1 was then preceded with molecular dynamic simulation for 15 ns to determine the stability of the model using TIP3P solvent model. Based on the simulation result, the backbone of human COX-1 showed deviation up to 2.25 Å and it showed a stable conformation after 6 ns of molecular dynamics run. This proved that the model was good enough to be used for molecular docking study.

Figure 2: Pharmacophore hypotheses for mPGES-1 that had been generated using LigandScout. Hypo4 comprised of five HY features (yellow sphere), two HBA features (red sphere) and a HBD feature (green sphere).
For human COX-2 enzyme, as of April 2018, there were seven (7) complex structures of human COX-2 available in PDB. Among these, complex structure with PDB ID: 5F19 was chosen to be used in this study as it had the highest resolution number (2.04 Å). 5F19 was bounded with aspirin and it inhibited COX-2 by acetylation of Ser530. The acetylation of Ser530 was observed in this complex structure. Hence, prior to protein preparation, the modified residue (OAS) at position 530 amino acid was changed to its mother residue (SER). Selective COX-2 inhibitor, celecoxib was used as the reference to validate docking protocol. The binding mode of the celecoxib in complexed with murine COX-2 (PDB ID: 3LN1) was compared with the re-docked celecoxib in human COX-2 (PDB ID: 5F19). The superimposed model of both celecoxib showed that the re-docked celecoxib had similar binding pose with the bounded celecoxib resulting in a low RMSD value (RMSD=0.442). This suggested that the docking protocol was suitable to be used in the study.
Virtual screening of COX inhibition
Similarity search was used to find chalcone derivatives in ASINEX database. From 523, 376 of compounds that were retrieved from ASINEX database, 392 of the compounds were chalcones and were subjected to virtual screening. The first stage of screening for potential COX-2 inhibitors was conducted using structurebased pharmacophore model. The pharmacophore model (phore 1) was developed based on known selective COX-2 inhibitors in complexed with COX-2 enzyme. Out of the 392 compounds, 42 hit compounds were mapped on the “phore 1” pharmacophore model with pharmacophore-fit values more than 45.00. The hit compounds were then cross screened with docking using GLIDE software for the evaluation of their binding orientations within COX-2 active site as well as their selectivity towards COX-2 enzyme.
The hit compounds were docked with both human COX-1 and COX-2 using XP mode of GLIDE docking. GLIDE software was used for primary screening of COX-2 inhibitor due to the characteristics of COX active site. COX active site contained highly hydrophobic area especially in the conserved region of COX active site. GLIDE with XP mode had produced consistent and accurate prediction in scoring or ranking compared to GOLD software with regards to hydrophobic binding sites [39-42]. GLIDE docking score is presented in docking energy score (kcal/mol); whereby a lower docking score shows better binding interactions. The criteria for selecting the specific COX- 2 inhibitors were; (a) compounds only docked into human COX-2 active site, or (b) compounds had COX-2 docking score lower than COX-1, and (c) the docking score for COX-2 were less than -8.0 kcal/ mol, and (d) the binding mode of the compound was also assessed to occupy the side pocket of COX-2. As a result, 15 hit compounds were selected for subsequent screening of mPGES-1 (Table 4). The list of COX-2 inhibitors with their structures, pharmacophore-fit score, docking score and binding interaction are presented in Table 4.
| No. | IDNUMBER | Pharmacophore-Fit Score | Docking score (kcal/mol) | |
|---|---|---|---|---|
| hCOX-1 | hCOX-2 | |||
| 1. | BAS 00127074 | 45.81 | ND | -8.002 |
| 2. | BAS 00384673 | 46.23 | -4.226 | -8.342 |
| 3. | BAS 00428711 | 45.75 | ND | -10.412 |
| 4. | BAS 00547888 | 44.77 | ND | -8.98 |
| 5. | BAS 00643043 | 45.46 | ND | -9.208 |
| 6. | BAS 00643060 | 45.06 | ND | -9.142 |
| 7. | BAS 00643061 | 55.46 | ND | -8.925 |
| 8. | BAS 00654798 | 46.72 | 2.983 | -8.464 |
| 9. | BAS 00791751 | 46.56 | ND | -10.61 |
| 10. | BAS 01121975 | 55.49 | -3.79 | -9.65 |
| 11. | BAS 01121978 | 47.26 | -8.753 | -10.029 |
| 12. | BAS 01316535 | 45.35 | -4.902 | -8.149 |
| 13. | BAS 01316573 | 45.28 | -7.842 | -8.852 |
| 14. | BAS 02332476 | 47.68 | ND | -9.207 |
| 15. | BAS 02557914 | 45.72 | -3.182 | -8.726 |
Table 4: Pharmacophore fit score and docking score for the hit compounds after screening for COX-2 inhibition.
mPGES-1 pharmacophore model generation and theoretical validation
To identify mPGES-1 inhibitors from the ASINEX database, a similar screening method that was used for COX-2 were adopted. Pharmacophore screening and subsequently molecular docking for mPGES-1 was conducted using 4BPM as crystal structure of mPGES-1. The structure-based pharmacophore model for mPGES-1 was developed using LigandScout based on protein-ligand complex of 5BQI as shown in Figure 2. The crystal structure of mPGES-1 was bounded with the most potent and non-acidic mPGES-1 inhibitor (4UL). The pharmacophore model of mPGES-1 comprised of five HY features, two HBA features and a HBD feature. The pharmacophore model was validated using EF. The relative EF for the pharmacophore model of mPGES-1 was 0.435 with the minimum features being set to 5 (Table 5). This pharmacophore model maps 100% of mPGES-1 inhibitors with 65% of false positive results.
| Pharmacophore hypothesis | n° of features | Chemical features | Min n° of required features | EF | Rel EF | Actives found (%) (n=10) | Decoys found (%) (n=21) |
|---|---|---|---|---|---|---|---|
| Hypothesis 4 (Hypo4) | 8 | 5 HY, 2 HBA, 1 HBD | 8 | 3.10 | 1.000 | 0.0 | 0 |
| 7 | 3.10 | 1.000 | 20.0 | 0 | |||
| 6 | 3.10 | 1.000 | 60.0 | 0 | |||
| 5 | 1.23 | 0.435 | 100.0 | 65 |
Table 5: The structure-based pharmacophore hypothesis generation for mPGES-1.
mPGES-1 structural analysis
mPGES-1 is an inducible, glutathione (GSH) dependent enzyme that is responsible for PGH2 conversion to inducible PGE2. It is formed as homotrimer with three equivalent active site cavities between each monomer interface and it is located on the endoplasmic reticulum. The binding site of mPGES-1 consists of large hydrophobic region and polar region [43]. mPGES-1 inhibitors can either occupy the binding site of the substrate PGH2 or partially displacing GSH [31]. Based on the study by Gupta et al. [9], several residues had been classified as important residues that played roles in interactions with inhibitors of different classes/scaffolds such as Arg70, Arg73, Asn74, Glu77, His113, Tyr117, Arg126, Ser127, Tyr130, Thr131 and Ala138. A potent inhibitor of mPGES-1 should make π-π interaction and/or polar/H-bond interactions with either Arg73, Asn74, Tyr117, Arg126, Tyr130 and/or Thr131 which replaced the mPGES-1 cofactor, GSH [9]. A compound that can occupy in both inhibitory sites (substrate and GSH binding site) is highly potent inhibitor for mPGES-1.
GSH is a cofactor for mPGES-1 protein that binds at the deeper part of polar region of mPGES-1 active site. Two terminal carboxylic groups of GSH have strong interactions with Arg38 and Arg73 which give GSH U-shape in the deeper part of the binding site [32]. Another residue that plays roles in GSH interaction is Tyr130, where it involves a π-stacking interaction and its glutamate side chain bound through a gamma linkage to the cysteine in GSH [32]. Therefore, mPGES-1 inhibitors should form π-π interaction or/and polar/H-bond interactions with these residues. There is another binding site for an inhibitor to bind, where PGH2 (substrate) binds and it is located between GSH binding sites. The binding site comprises of aromatic (Phe44, His53) and polar (Arg52) residues at the apex of the binding site that is near to GSH binding site. It is extended until aliphatic (Pro124, Val128, Leu132) and polar (Ser127, Thr131) residues. A potent mPGES-1 inhibitor could form π-interaction and/or polar/Hbond interaction in this binding site as seen for 4UL, 7DN and LVJ; the inhibitors co-crystallized with GSH and mPGES-1 structure (5BQI, 5TL9 and 4BPM respectively). Based on the study by He and Lai [31], mPGES-1 inhibitors could bind to substrate and GSH binding site simultaneously. Therefore, the docking study was performed in the presence and absence of GSH in the binding site of mPGES-1.
Virtual screening for mPGES-1 inhibition
Fifteen (15) compounds from virtual screening of COX-2 were screened for inhibition of mPGES-1 in a similar manner as inhibition of COX-2. The compounds were firstly screened using pharmacophore model of mPGES-1 and then proceeded with molecular docking using 4BPM as crystal structure of mPGES-1. Based on the pharmacophore screening, seven (7) out of 15 compounds were selected for the second stage of screening. From seven (7) compounds, only six (6) compounds were docked to the active site of mPGES-1. The results for pharmacophore screening and docking are presented in Table 6. Finally, the binding modes of mPGES-1 for only the top three compounds were reported. The hit compounds which can act as dual-target inhibitors; COX-2 inhibitors and mPGES-1 inhibitors are known as BAS00384673, BAS00643043 and BAS02557914. Therefore, the binding modes of these compounds at the binding sites were further analyzed to understand the molecular interactions of the hit compounds with both enzymes.
| No. | 2D pharmacophore mapping using “phore 1” | Fit Score | 2D ligand interaction diagram of mPGES-1 with GSH/Docking score (kcal/mol) | 2D ligand interaction diagram of mPGES-1 without GSH/Docking score (kcal/mol) | ||
|---|---|---|---|---|---|---|
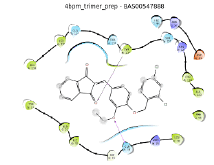
| 1 | BAS 00384673 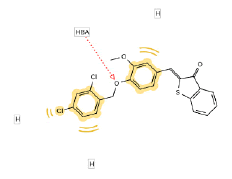 |
48.44 |  |
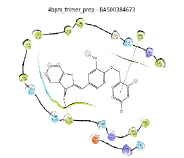 |
||
| -6.967 | -6.109 | |||||
| 2 | BAS 00547888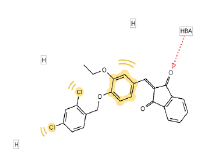 |
45.60 | 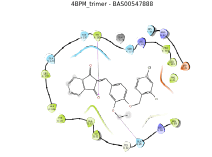 |
 |
||
| -7.146 | -6.109 | |||||
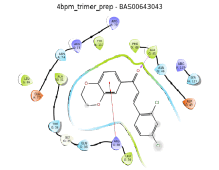
| 3 | BAS 00643043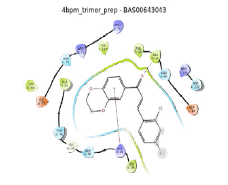 |
55.50 | 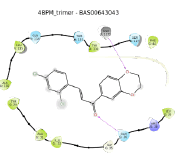 |
 |
||
| -5.139 | -4.696 | |||||
| 4 | BAS 01121975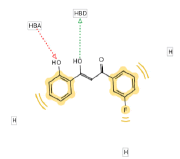 |
56.37 | 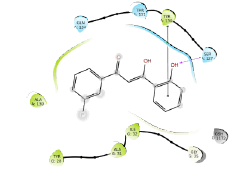 |
 |
||
| -6.437 | -7.039 | |||||
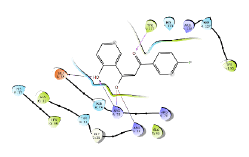
| 5 | BAS 01121978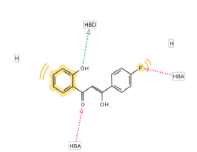 |
56.13 | 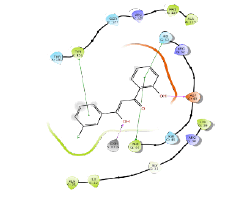 |
 |
||
| -6.258 | -4.285 | |||||
| 6 | BAS 02557914 |
54.98 |  |
 |
||
| -5.611 | -5.459 | |||||
Table 6: Final hit compounds after mPGES-1 screening.
Analysis of binding interactions of the hit compounds in COX and mPGES-1 binding sites
In docking analysis, compound BAS00384673 was found to occupy the hydrophobic region and extended to lobby region of the hCOX binding site (Figure 3). The benzo(b)thiophe-3(2H)-one ring resided in the hydrophobic region and the ring formed hydrophobic interaction with residues in the hydrophobic region of both hCOX binding sites (Val349, Leu352, Tyr385, Trp387, Val523/Ile523 and Ala527). However, the benzo(b)thiophe-3(2H)-one ring possessed different binding pose in both hCOX binding sites where the ketone group was pointed towards Ala527 in hCOX-1 and towards Ser530 in hCOX-2 binding site. The benzene ring with O-methyl group formed π-π interaction with the benzene ring of Tyr355 at the entrance of both COX-1 and COX-2 binding site. Furthermore, compound BAS00384673 had different binding orientation especially for the dichlorobenzene group which resided in the lobby region of the both COX-1 and COX-2 binding sites. In COX-1 binding site, the group formed hydrophobic interaction with Thr89 and Leu93 at the lobby region while the group bends toward Arg120 and the benzene ring of group made a π-π interaction with guanidinium group of Arg120 in COX-2 binding site.

Figure 3: Different binding mode of BAS00384673 in COX binding site (a) hCOX-1 and (b) hCOX-2. Compound in the active site were represented in sticks (coloured by atom types: C grey, O red, N blue, polar, H light grey, Cl green). H-bonds were represented in orange dotted line.
Binding modes of BAS00384673 in the mPGES-1 without GSH and with GSH are shown in Figure 4. Figure 4a shows the binding mode of BAS0384673 in the absence of GSH, where the methoxybenzene oxygen formed a hydrogen bond with amide group of A: Gln36 and the side chain OH of B: Tyr130 formed a hydrogen bond with the benzo(b) thiophe-3(2H)-one ketone. Moreover, the benzo(b) thiophe-3(2H)-one ring were rested in the hydrophobic cleft toward the external binding groove of mPGES-1 which comprised of A: Tyr28, A: Ala31, A: Ile32, A: Gln36, B: Tyr130, B: Thr131, B: Gln134, B: Leu135 and B: Ala138. The benzene ring with chlorine substituent at ortho- and para- position of compounds BAS00384673 were stabilized by hydrophobic interaction with A: Leu39 and B: Pro120, respectively. Additionally, the ring also formed edge-toface π-π interaction with A: Phe44 which was one of the important residues in mPGES-1 activity. In the presence of GSH (Figure 4b), although the compound had the same binding orientation as in the mPGES-1 without GSH; it only had a hydrogen bond with amide side chain of A: Gln36.

Figure 4: Different binding mode of BAS00384673 in mPGES-1 binding site (represented in ribbons, pink for Chain A, blue for Chain B) in the (a) absence and (b) presence of the cofactor (GSH). Residues in the active site were represented in sticks (colored by atom types: C orange, O red, N blue, polar, H light grey, GSH green). H-bonds were represented in yellow/orange dotted line, while π-π interactions were depicted with cyan dotted lines.
In the case of BAS00643043, the compound was able to dock into COX-2 binding site by occupying the side pocket and the hydrophobic regions of binding site with docking score of -9.028 kcal/mol (Figure 5). The thiazole ring of BAS00643043 was inserted into the side pocket region of hCOX-2 binding site and the oxygen atom of the thiazole ring formed hydrogen bond with His90. Plus, the thiazole ring also had established hydrophobic interaction with Ala516, Phe518 and Val523 while the dichlorobenzene ring was protruded towards hydrophobic region of the hCOX-2 binding site and formed hydrophobic interaction with Phe381, Leu384, Trp387, Val523, Ala527 and Ser530. The chlorine (Cl) atom at para position also formed a π interaction with Tyr385 and another Cl atom at ortho position formed a halogen bond with Ser530.

Figure 5: Binding mode of BAS00643043 in hCOX-2 binding site. Compound BAS00643043 are represented in line (colored by atom types: C grey, O red, N blue, polar, H light grey, Cl green). H-bonds are represented in orange dotted line.
BAS00643043 bond to the binding site of mPGES-1 in different orientations, in the presence and absence of GSH (Figure 6). In the absence of GSH, the compound complemented the surface of the binding site above the hydrophobic cleft (above B: Tyr130) and moved towards the glutathione binding site as the ketone group pointed toward GSH binding site, and thus formed a “L” shape interaction. The 1-4-benzodoxan ring was orientated towards hydrophobic cleft and it formed a stable π-cation interaction with guanidinium of A: Arg38. Meanwhile, the benzene ring with chlorine substituent occupied the substrate binding site above the cleft, where the chlorine at ortho-position formed hydrophobic interaction with B: Ser127 and A: Asp49 and chlorine at para-position formed hydrophobic interaction with A: Phe44 and A: Leu39. While for the binding mode of the compound in the presence of GSH; the benzene with chlorine substituent was embedded in a hydrophobic cleft. The hydrophobic cleft was formed by mainly aromatic or hydrophobic amino acid (B: Thr131, B: Gln134, B: Leu135, B: Ala138, A: Tyr28, A: Ala31). Thus, a hydrogen bond was formed between ketone group of the compound and amide group of A: Gln36.

Figure 6: Different binding modes of BAS00643043 in the binding site of mPGES-1 (represented in ribbons, pink for Chain A, blue for Chain B) in the (a) absence and (b) presence of the cofactor (GSH). Residues in the active site are represented in sticks (colored by atom types: C orange, O red, N blue, polar, H light grey, GSH green). H-bonds are represented in yellow/orange dotted line, while π-π interactions are depicted with cyan dotted lines.
BAS02557914 presents in trans-conformation in COX-1 and cis-conformation in COX-2 (Figure 7). Ring A of BAS02557914 has a O-methyl group at para position which is strengthened by hydrophobic interactions with Val349, Leu352, Ile523/Val523 and Ala527 in hydrophobic region of the binding sites of both COX; however the binding pose varied in the binding sites for different COX. The ring B of BAS02557914 formed π-π interaction with guanidinium group of Arg120 and the thiazole substituent at para position of ring B established hydrophobic interaction with Thr89 and Leu93 at the lobby region of COX-1 binding site. In comparison, the ring B of BAS02557914 was stabilized by hydrophobic interaction with Val89, Leu93 and Tyr355 at lobby region of COX-2 binding site. Besides, the thiazole ring was oriented towards Arg120 and formed hydrophobic interaction with Arg120 and Leu123.

Figure 7: Different binding mode of BAS02557914 in (a) hCOX-1 and (b) hCOX-2 binding site. Compound BAS0257914 is represented in line (coloured by atom types: C grey, O red, N blue, polar, H light grey, Cl green). H-bonds are represented in orange dotted line.
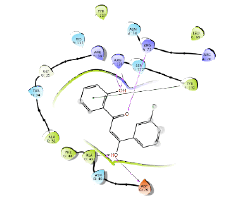
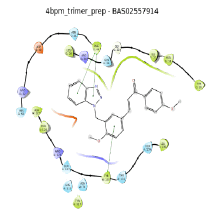
On the other hand, in the absence of GSH (Figure 8a), the thiazole ring of BAS02557914 was inserted into a groove above GSH at the top pocket and formed two π-π interaction with benzene ring of A: Phe44; and hydrophobic interaction with A: Leu39, B: Ala123 and B: Pro124. In addition, the core methoxybenzene which was near to GSH binding site, formed a π-π interaction with benzene ring of B:Tyr130 and polar interaction with B: Arg126. Meanwhile, the “tail” of methoxybenzene group of BAS02557914 formed hydrophobic interaction with A: Ile32, A: Ala31, A: Tyr28 and B: Leu135 at the hydrophobic cleft of the binding site of mPGES-1. In the case of GSH (Figure 8b), BAS02557914 is in its cis conformer, where the methoxybenzene group at the “tail” was directed towards the hydrophobic cleft and has hydrophobic interaction with B: Tyr120 and A: Ile32. In addition, the ketone group formed a polar interaction with the amide group of A: Gln36; and the thiazole ring bent and formed a “U” shape and have a stable π-π interaction with A: His53.

Figure 8: Different binding mode of BAS02557914 in the binding sites of mPGES-1 (represented in ribbons, pink for Chain A, blue for Chain B) in the (a) absence and (b) presence of the cofactor (GSH). Residues in the active site are represented in sticks (colored by atom types: C orange, O red, N blue, polar, H light grey, GSH green). H-bonds are represented in yellow/orange dotted line, while π-π interactions are depicted with cyan dotted lines.
MM-GBSA calculation
The top three hit compounds that were successfully docked to COX and mPGES-1 were further studied with MM-GBSA calculation as the post-docking filter and to identify the inhibitor potential. Compounds BAS00384673, BAS00643043 and BAS02557914 were used as input for MM-GBSA analysis and the binding poses of each of the compound in COX and mPGES-1 targets were used for the calculation. According to Table 7, the binding free energies (ΔG bind) of the BAS00384673-COX-1, BAS00384673-COX-2, BAS00384673- mPGES-1 (with GSH), BAS00384673-mPGES-1 (without GSH), BAS00643043-COX-1, BAS00643043-COX-2, BAS00643043- mPGES-1 (with GSH), BAS00643043-mPGES-1 (without GSH), BAS02557914-COX-1, BAS02557914-COX-2, BAS02557914- mPGES-1 (with GSH), and BAS02557914-mPGES-1 (without GSH) were in agreement with the docking scores. This shows that the binding abilities of these compounds towards COX-2 are greater than to COX-1 and mPGES-1.
| Compounds | ΔG bind (kcal/mol) | |||
|---|---|---|---|---|
| COX-1 | COX-2 | mPGES-1 | ||
| with GSH | without GSH | |||
| BAS00384673 | -85.359 | -90.005 | -58.388 | -49.893 |
| BAS00643043 | - | -60.470 | -46.014 | -43.683 |
| BAS02557914 | -61.726 | -69.779 | -46.710 | -46.630 |
Table 7: Prime MM-GBSA energy for BAS00384673, BAS00643043 and BAS0255791 within COX and mPGES-1.
Conclusion
In this work, a high-throughput in silico screening of ASINEX database was performed by adopting structure-based approach using LigandScout and molecular docking by GLIDE to obtain selective hit compounds that can inhibit both COX-2 and mPGES-1. A diverse pharmacophore models were generated for COX-2 and were able to recognize the different chemical scaffold of COX-2. The pharmacophore model was built by hybrid pharmacophore based on five (5) COX- 2 inhibitors and the best hypothesis was named as “phore 1”. After the pharmacophore screening, molecular docking was carried out to improve the sensitivity and specificity of choosing the hit compounds. In the case of COX-2 screening, both human COX (COX-1 and COX- 2) crystal structures were used to assist in the evaluation of conserved interactions with the key amino acids at the active site. Fifteen (15) hit compounds of chalcone scaffold were identified as COX-2 inhibitors and these hit compounds were further screened for mPGES-1 using similar modelling approach for COX-2; pharmacophore screening and docking analysis. As for mPGES-1 pharmacophore, a complex mPGES-1 structure bounded with a potent mPGES-1 inhibitor was used to build a structure-based pharmacophore for mPGES-1 model. Using this method, three lead compounds were successfully identified as selective dual-inhibitor of COX-2 and mPGES-1; BAS00384673, BAS00643043, and BAS02557914. It is noteworthy that these three compounds have potential to become anti-inflammatory drug with less undesirable side effects.
Acknowledgements
The authors were thankful to Integrative Pharmacogenomics Institute (iPROMISE), Universiti Teknologi MARA (UiTM), Malaysia for providing the facilities to carry out this study. This study was supported by the Transdisciplinary Research Grant Scheme (TRGS) from the Ministry of Higher Education (Grant No.: 600-RMI/TRGS 5/3 (1/2014)).
References
- Simmons DL, Botting RM, Hla T (2004) Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387-437.
- Botting RM (2006) Inhibitors of cyclooxygenases: Mechanisms, selectivity and uses. J Physiol Pharmacol 57: 113-124.
- Crofford LJ (2013) Use of NSAIDs in treating patients with arthritis. Arthritis Res Ther 15: S2.
- Rao PNP, Knaus EE (2008) Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci 11: 81-110.
- Hawkey CJ (1999) New drug classes - COX-2 inhibitors. Lancet 353: 307-314.
- Chandrasekharan NV, Simmons DL (2004) The cyclooxygenases. Genome Biol 5: 241.
- Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, et al. (2013) Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther 139: 189-212.
- Wendell SG, Baffi C, Holguin F (2014) Fatty acids, inflammation, and asthma. J Allergy Clin Immunol 133: 1255-1264.
- Gupta A, Chaudhary N, Kakularam KR, Pallu R, Polamarasetty A (2015) The augmenting effects of Desolvation and Conformational energy terms on the predictions of docking programs against mPGES-1. PLoS One 10: e0134472
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, et al. (2006) Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest 116: 1391-1399.
- Kamal A, Mallareddy A, Suresh P, Lakshma NV, Shetti RV, et al. (2012) Synthesis and anticancer activity of 4β-alkylamidochalcone and 4β-cinnamido linked podophyllotoxins as apoptotic inducing agents. Eur J Med Chem 47: 530-545.
- Sashidhara KV, Kumar A, Kumar M, Sarkar J, Sinha S (2010) Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg Med Chem Lett 20: 7205-7211.
- Trivedi JC, Bariwal JB, Upadhyay KD, Naliapara YT, Joshi SK, et al. (2007) Improved and rapid synthesis of new coumarinyl chalcone derivatives and their antiviral activity. Tetrahedron Lett 48: 8472-8474.
- Gupta D, Jain DK (2015) Chalcone derivatives as potential antifungal agents: Synthesis, and antifungal activity. J Adv Pharm Technol Res 6: 114-117.
- Siddiqui ZN, Musthafa TN, Ahmad A, Khan AU (2011) Thermal solvent-free synthesis of novel pyrazolyl chalcones and pyrazolines as potential antimicrobial agents. Bioorganic Med Chem Lett 21: 2860-2865.
- Yadav N, Dixit SK, Bhattacharya A, Mishra LC, Sharma M, et al. (2012) Antimalarial activity of newly synthesized chalcone derivatives in vitro. Chem Biol Drug Des 80: 340-347.
- Awasthi SK, Mishra N, Kumar B, Sharma M, Bhattacharya A, et al. (2009) Potent antimalarial activity of newly synthesized substituted chalcone analogs in vitro. Med Chem Res 18: 407-420.
- Enoki T, Ohnogi H, Nagamine K, Kudo Y, Sugiyama K, et al. (2007) Antidiabetic activities of chalcones isolated from a Japanese herb, Angelica keiskei. J Agric Food Chem 55: 6013-6017.
- Doan TN, Tran DT (2011) Synthesis, antioxidant and antimicrobial activities of a novel series of chalcones, pyrazolic chalcones and allylic chalcones. Pharmacol Pharm 2: 282-288.
- Chen YH, Wang WH, Wang YH, Lin ZY, Wen CC, et al. (2013) Evaluation of the anti-inflammatory effect of chalcone and chalcone analogues in a Zebrafish model. Molecules 18: 2052-2060.
- Liu Z, Tang L, Zou P, Zhang Y, Wang Z, et al. (2014) Synthesis and biological evaluation of allylated and prenylated mono-carbonyl analogs of curcumin as anti-inflammatory agents. Eur J Med Chem 74: 671-682.
- Won SJ, Liu CT, Tsao LT, Weng JR, Ko HH, et al. (2005) Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur J Med Chem 40: 103-112.
- Singh H, Sidhu S, Chopra K, Khan MU (2016) Hepatoprotective effect of trans-Chalcone on experimentally induced hepatic injury in rats: inhibition of hepatic inflammation and fibrosis. Can J Physiol Pharmacol 94: 879-887.
- Rofiee MS, Yusof MI, Hisam EEA, Bannur Z, Zakaria ZA, et al. (2015) Isolating the metabolic pathways involved in the hepatoprotective effect of Muntingia calabura against CCl4-induced liver injury using LC/MS Q-TOF. J Ethnopharmacol 166: 109-118.
- Yusof MIM, Salleh MZ, Kek TL, Ahmat N, Azmin NFN, et al. (2013) Activity-guided isolation of bioactive constituents with antinociceptive activity from Muntingia calabura L. leaves using the formalin test. Evidence-based Complement Altern Med 1-27.
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, et al. (1996) Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384: 644-648.
- Limongelli V, Bonomi M, Marinelli L, Gervasio FL, Cavalli A, et al. (2010) Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc Natl Acad Sci USA 107: 5411-5416.
- Rocca R, Costa G, Artese A, Parrotta L, Ortuso F, et al. (2016) Hit identification of a novel dual binder for h-telo/c-myc G-Quadruplex by a combination of pharmacophore structure-based virtual screening and docking refinement. ChemMedChem 11: 1721-1733.
- Ibrahim TM, Bauer MR, Dörr A, Veyisoglu E, Boeckler FM (2015) pROC-Chemotype plots enhance the interpretability of benchmarking results in structure-based virtual screening. J Chem Inf Model 55: 2297-2307.
- Dhanjal JK, Sreenidhi AK, Bafna K, Katiyar SP, Goyal S, et al. (2015) Computational structure-based de novo design of hypothetical inhibitors against the anti-inflammatory target COX-2. PLoS One 10: e0134691.
- He S, Lai L (2011) Molecular docking and competitive binding study discovered different binding modes of microsomal prostaglandin E synthase-1 inhibitors. J Chem Inf Model 51: 3254-3261.
- Lauro G, Tortorella P, Bertamino A, Ostacolo C, Koeberle A, et al. (2016) Structure-based design of microsomal prostaglandin E2 synthase-1 (mPGES-1) inhibitors using a virtual fragment growing optimization scheme. Chem Med Chem 11: 612-619.
- Windsor MA, Valk PL, Xu S, Banerjee S, Marnett LJ (2013) Exploring the molecular determinants of substrate-selective inhibition of cyclooxygenase-2 by lumiracoxib. Bioorg Med Chem Lett 23: 5860–5864.
- Wang JL, Limburg D, Graneto MJ, Springer J, Hamper JR, et al. (2010) The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorganic Med Chem Lett 20: 7159-7163.
- Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, et al. (2003) A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem 278: 45763-45769.
- Epstein CJ, Goldberger RF, Anfinsen CB (1963) The genetic control of tertiary protein structure: Studies with model systems. Cold Spring Harb Symp Quant Biol 28: 439-449.
- Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. EMBO J 5: 823-826.
- Fiser A (2010) Template-based protein structure modelling. Methods Mol Biol 673: 73-94.
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739-1749.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, et al. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49: 6177-6196.
- Perola E, Walters WP, Charifson PS (2004) A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 56: 235-249.
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20: 13384-13421.
- Sjogren T, Nord J, Ek M, Johansson P, Liu G et al. (2013) Crystal structure of microsomal prostaglandin E2 synthase provides insight into diversity in the MAPEG superfamily. Proc Natl Acad Sci USA 110: 3806-3811.